
Whole-Genome Sequencing and Annotation of the Yeast Clavispora santaluciae Reveals Important Insights about Its Adaptation to the Vineyard Environment

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Sample Collection, and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation
2.4. Homology Analysis, Comparative Genomics, and Phylogenomics
3. Results and Discussion
3.1. Sequencing, De Novo Assembly, and Annotation of Clavispora Santaluciae Genome
3.2. Comparative Genomics of Clavispora Santalucieae Strains
3.3. Functional Annotation of Clavispora Santaluciae Proteome
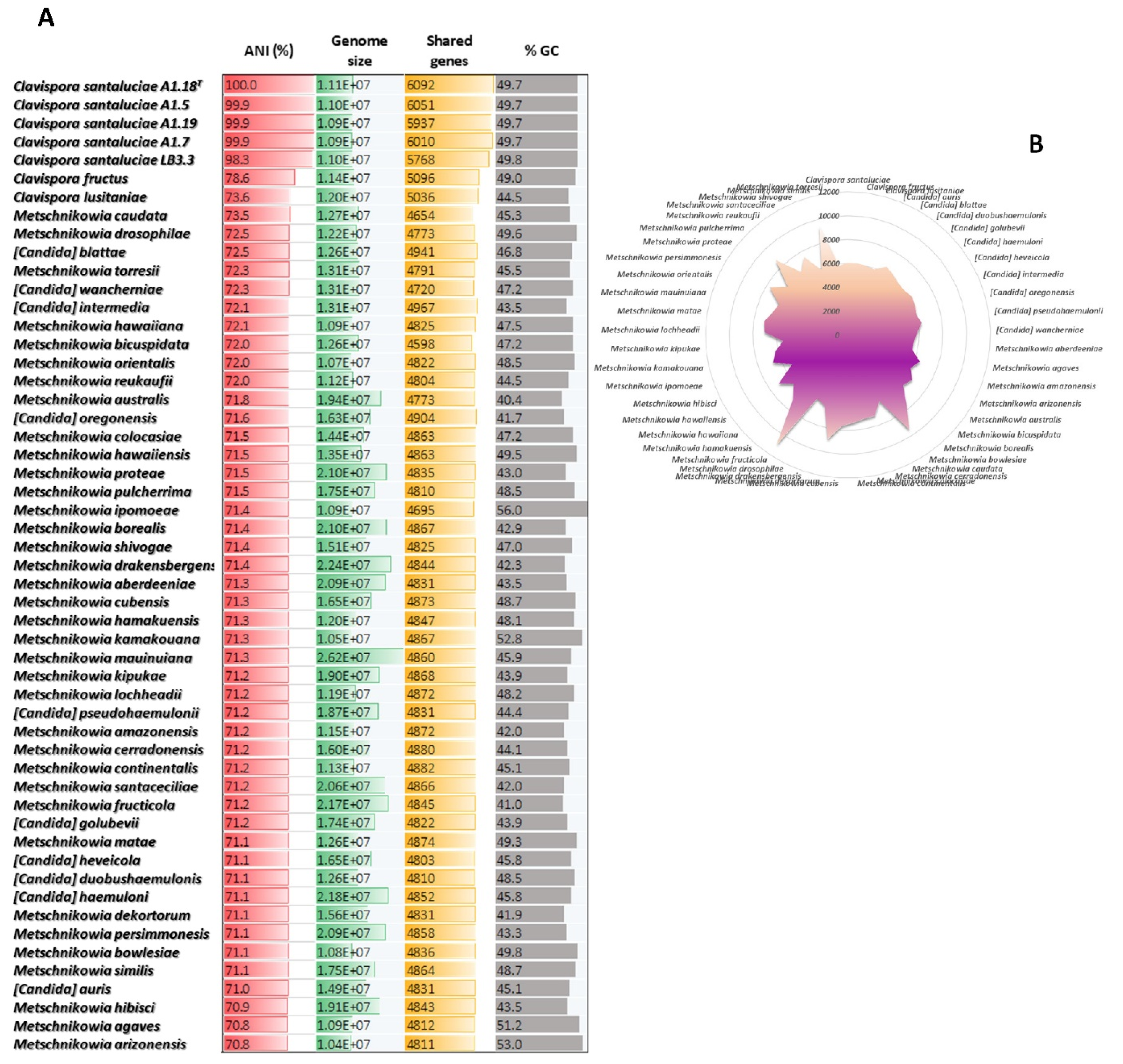
3.4. Interspecific Genomic Variability of Metschnikowiaceae
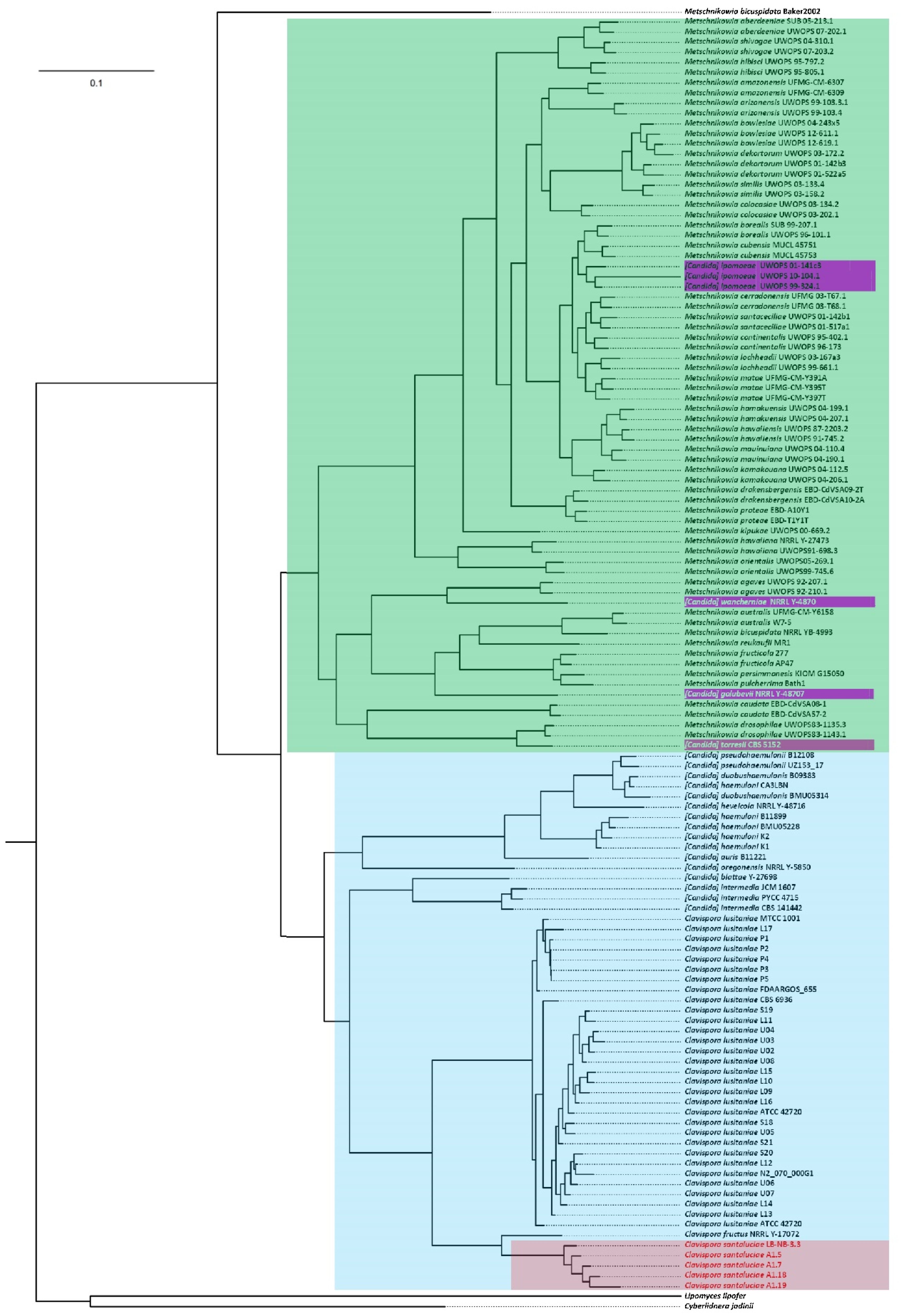
3.5. Phylogenomics of Metschnikowiacea
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drumonde-Neves, J.; Franco-Duarte, R.; Lima, T.; Schuller, D.; Pais, C. Association between grape yeast communities and the vineyard ecosystems. PLoS ONE 2017, 12, e0169883. [Google Scholar] [CrossRef] [PubMed]
- Drumonde-Neves, J.; Franco-Duarte, R.; Lima, T.; Schuller, D.; Pais, C. Yeast biodiversity in vineyard environments is increased by human intervention. PLoS ONE 2016, 11, e0160579. [Google Scholar] [CrossRef] [Green Version]
- Drumonde-Neves, J.; Franco-Duarte, R.; Vieira, E.; Mendes, I.; Lima, T.; Schuller, D.; Pais, C. Differentiation of Saccharomyces cerevisiae populations from vineyards of the Azores Archipelago: Geography vs. Ecology. Food Microbiol. 2018, 74, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Drumonde-Neves, J.; Čadež, N.; Domínguez, Y.R.; Gallmetzer, A.; Schuller#, D.; Lima, T.; Pais, C.; Franco-Duarte, R. Clavispora santaluciae f.a., sp. nov., a novel ascomycetous yeast species isolated from grapes. Int. J. Syst. Evol. Microbiol. 2020, 70, 6307–6312. [Google Scholar] [CrossRef]
- Gárdonyi, M.A.; Österberg, M.A.; Rodrigues, C.; Spencer-Martins, I.; Hahn-Hägerdal, B. High capacity xylose transport in Candida intermedia PYCC 4715. FEMS Yeast Res. 2003, 3, 45–52. [Google Scholar] [CrossRef]
- Geijer, C.; Faria-Oliveira, F.; Moreno, A.D.; Stenberg, S.; Mazurkewich, S.; Olsson, L. Genomic and transcriptomic analysis of Candida intermedia reveals the genetic determinants for its xylose-converting capacity. Biotechnol. Biofuels 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.D.; Tomás-Pejó, E.; Olsson, L.; Geijer, C. Candida intermedia CBS 141442: A novel glucose/xylose co-fermenting isolate for lignocellulosic bioethanol production. Energies 2020, 13, 5363. [Google Scholar] [CrossRef]
- Drumonde-Neves, J.; Fernandes, T.; Lima, T.; Pais, C.; Franco-Duarte, R. Learning from 80 years of studies: A comprehensive catalogue of non-Saccharomyces yeasts associated with viticulture and winemaking. FEMS Yeast Res. 2021, 21, foab017. [Google Scholar] [CrossRef]
- Mingorance-Cazorla, L.; Clemente-Jiménez, J.M.; Martínez-Rodríguez, S.; Las Heras-Vázquez, F.J.; Rodríguez-Vico, F. Contribution of different natural yeasts to the aroma of two alcoholic beverages. World J. Microbiol. Biotechnol. 2003, 19, 297–304. [Google Scholar] [CrossRef]
- Morata, A.; Loira, I.; Escott, C.; del Fresno, J.M.; Bañuelos, M.A.; Suárez-Lepe, J.A. Applications of Metschnikowia pulcherrima in wine biotechnology. Fermentation 2019, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Daniel, H.M.; Lachance, M.A.; Kurtzman, C.P. On the reclassification of species assigned to Candida and other anamorphic ascomycetous yeast genera based on phylogenetic circumscription. Antonie Leeuwenhoek 2014, 106, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, C.P.; Robnett, C.J.; Basehoar, E.; Ward, T.J. Four new species of Metschnikowia and the transfer of seven Candida species to Metschnikowia and Clavispora as new combinations. Antonie Leeuwenhoek 2018, 111, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Lachance, M.-A.; Hurtado, E.; Hsiang, T. A stable phylogeny of the large-spored Metschnikowia clade. Yeast 2016, 33, 261–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.X.; Opulente, D.A.; Kominek, J.; Zhou, X.; Steenwyk, J.L.; Buh, K.V.; Haase, M.A.B.; Wisecaver, J.H.; Wang, M.; Doering, D.T.; et al. Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 2018, 175, 1533–1545.e20. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, K.; Sherlock, G. Preparation of yeast DNA sequencing libraries. Cold Spring Harb. Protoc. 2016, 2016, 871–876. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 2020, 16, e1007981. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef] [Green Version]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Weib, C.L.; Pais, M.; Cano, L.M.; Kamoun, S.; Burbano, H.A. nQuire: A statistical framework for ploidy estimation using next generation sequencing. BMC Bioinform. 2018, 19, 122. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. ISBN 9781493991730. [Google Scholar]
- Yoon, S.-H.; Ha, S.-M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Lu, Z.; Wang, S.; Jing-Yan Wang, J.; Gao, X. CMsearch: Simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016, 32, i332–i340. [Google Scholar] [CrossRef]
- Arias-Carrasco, R.; Vásquez-Morán, Y.; Nakaya, H.I.; Maracaja-Coutinho, V. StructRNAfinder: An automated pipeline and web server for RNA families prediction. BMC Bioinform. 2018, 19, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.I.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, C.; Rito, T.; Vieira, D.; Fernandes, T.; Pais, C.; Sousa, M.J.; Soares, P.; Franco-Duarte, R. Improvement of torulaspora delbrueckii genome annotation: Towards the exploitation of genomic features of a biotechnologically relevant yeast. J. Fungi 2021, 7, 287. [Google Scholar] [CrossRef]
- Sun, Y.B. FasParser: A package for manipulating sequence data. Zool. Res. 2017, 38, 110–112. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Durrens, P.; Klopp, C.; Biteau, N.; Fitton-Ouhabi, V.; Dementhon, K.; Accoceberry, I.; Sherman, D.J.; Noël, T. Genome Sequence of the Yeast Clavispora lusitaniae Type Strain CBS 6936. Genome Announc. 2017, 5, 30–31. [Google Scholar] [CrossRef] [Green Version]
- Kannan, A.; Asner, S.A.; Trachsel, E.; Kelly, S.; Parker, J.; Sanglard, D. Comparative Genomics for the Elucidation of Multidrug Resistance in Candida lusitaniae. MBio 2019, 10, e02512-19. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.D.; Tellgren-Roth, C.; Soler, L.; Dainat, J.; Olsson, L.; Geijer, C. Complete Genome Sequences of the Xylose-Fermenting Candida intermedia Strains CBS 141442 and PYCC 4715. Genome Announc. 2017, 5, e00138-17. [Google Scholar] [CrossRef] [Green Version]
- Garbarino, J.E.; Gibbons, I.R. Expression and genomic analysis of midasin, a novel and highly conserved AAA protein distantly related to dynein. BMC Genom. 2002, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Franco-Duarte, R.; Bigey, F.; Carreto, L.; Mendes, I.; Dequin, S.; Santos, M.A.S.; Pais, C.; Schuller, D. Intrastrain genomic and phenotypic variability of the commercial Saccharomyces cerevisiae strain Zymaflore VL1 reveals microevolutionary adaptation to vineyard environments. FEMS Yeast Res. 2015, 15, fov063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Duarte, R.; Umek, L.; Zupan, B.; Schuller, D. Computational approaches for the genetic and phenotypic characterization of a Saccharomyces cerevisiae wine yeast collection. Yeast 2009, 26, 675–692. [Google Scholar] [CrossRef] [Green Version]
- Franco-Duarte, R.; Umek, L.; Mendes, I.; Castro, C.C.; Fonseca, N.; Martins, R.; Silva-Ferreira, A.C.; Sampaio, P.; Pais, C.; Schuller, D. New integrative computational approaches unveil the Saccharomyces cerevisiae pheno-metabolomic fermentative profile and allow strain selection for winemaking. Food Chem. 2016, 211, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Davies, G.J.; Gloster, T.M.; Henrissat, B. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 2005, 15, 637–645. [Google Scholar] [CrossRef]
- Piombo, E.; Sela, N.; Wisniewski, M.; Hoffmann, M.; Gullino, M.L.; Allard, M.W.; Levin, E.; Spadaro, D.; Droby, S. Genome sequence, assembly and characterization of two Metschnikowia fructicola strains used as biocontrol agents of postharvest diseases. Front. Microbiol. 2018, 9, 593. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, T.; Silva-Sousa, F.; Pereira, F.; Rito, T.; Soares, P.; Franco-Duarte, R.; Sousa, M.J. Biotechnological Importance of Torulaspora delbrueckii: From the Obscurity to the Spotlight. J. Fungi 2021, 7, 712. [Google Scholar] [CrossRef]
- Sahay, S. Wine enzymes: Potential and practices. Enzym. Food Biotechnol. Prod. Appl. Futur. Prospect. 2018, 73–92. [Google Scholar] [CrossRef]
- Daenen, L.; Saison, D.; Sterckx, F.; Delvaux, F.R.; Verachtert, H.; Derdelinckx, G. Screening and evaluation of the glucoside hydrolase activity in Saccharomyces and Brettanomyces brewing yeasts. J. Appl. Microbiol. 2008, 104, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Opulente, D.A.; Kominek, J.; Shen, X.X.; Zhou, X.; Labella, A.L.; Bradley, N.P.; Eichman, B.F.; Čadež, N.; Libkind, D.; et al. Extensive loss of cell-cycle and DNA repair genes in an ancient lineage of bipolar budding yeasts. PLoS Biol. 2019, 17, e3000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; de Montigny, J.; Marck, C.; Neuvéglise, C.; Talla, E.; et al. Genome evolution in yeasts. Nature 2004, 430, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Scannell, D.R.; Butler, G.; Wolfe, K.H. Yeast genome evolution—the origin of the species. Yeast 2007, 24, 929–942. [Google Scholar] [CrossRef]
- Gómez, S.; Berdugo, S.; Mena, R. Occurrence of indigenous arbuscular mycorrhizal fungi associated with the rhizosphere of the naidí palm in Colombia. Cienc. Tecnol. Agropecu. 2020, 21, e1275. [Google Scholar]
- Restrepo-Correa, S.; Pineda-Meneses, E.; Rios-Osorio, L. Mechanisms of action of fungi AND bacteria used as biofertilizers in agricultural soils: A systematic review. Corpoica. Tecnol. Agropecu. 2017, 18, 335–351. [Google Scholar]
- Youdkes, D.; Helman, Y.; Burdman, S.; Matan, O.; Jurkevitch, E. Potential control of potato soft rot disease by the obligate predators bdellovibrio and like organisms. Appl. Environ. Microbiol. 2020, 86, e02543-19. [Google Scholar] [CrossRef] [PubMed]
- Leon-Ttacca, B.; Arévalo-Gardini, E.; Bouchon, A.S. Sudden death of Theobroma cacao L. caused by Verticillium dahliae Kleb. In Peru and its in vitro biocontrol. Cienc. Tecnol. Agropecu. 2019, 20, 133–148. [Google Scholar] [CrossRef]
- López-Hernández, F.; Cortés, A.J. Last-Generation Genome–Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.). Front. Genet. 2019, 10, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, M.W.; Cortés, A.J.; Farmer, A.D.; Huang, W.; Ambachew, D.; Varma Penmetsa, R.; Carrasquilla-Garcia, N.; Assefa, T.; Cannon, S.B. Uneven recombination rate and linkage disequilibrium across a reference SNP map for common bean (Phaseolus vulgaris L.). PLoS ONE 2018, 13, e0189597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.J.; López-Hernández, F.; Osorio-Rodriguez, D. Predicting Thermal Adaptation by Looking Into Populations’ Genomic Past. Front. Genet. 2020, 11, 564515. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A1.18T | A1.5 | A1.7 | A1.19 | LB-NB-3.3 | ||
|---|---|---|---|---|---|---|
| Canu assembler | Assembly length (bp) | 11,088,431 | 11,018,248 | 10,921,443 | 10,861,576 | 11,019,028 |
| Number of scaffolds | 43 | 13 | 46 | 86 | 30 | |
| N50 (bp) | 315,943 | 802,369 | 355,153 | 1,329,122 | 494,470 | |
| L50 | 7 | 3 | 6 | 14 | 7 | |
| Number of N’s per 100 Kb | 0 | 0 | 0 | 0 | 0 | |
| Number of scaffolds > 5000 bp | 29 | 11 | 28 | 53 | 23 | |
| Total length > 5000 bp | 10,780,215 | 10,974,595 | 10,557,056 | 10,118,395 | 10,856,688 | |
| Masurca assembler | Substitution errors revised | 42 | 8 | 141 | 428 | 70 |
| Insertion/Deletion errors revised | 1686 | 536 | 2825 | 6144 | 1607 | |
| Assembly length (bp) | 11,089,145 | 11,018,616 | 10,922,446 | 10,863,639 | 11,019,715 | |
| Number of scaffolds | 43 | 13 | 46 | 86 | 30 | |
| N50 (bp) | 532,329 | 1,048,728 | 654,799 | 218,696 | 650,701 | |
| L50 | 7 | 3 | 6 | 14 | 7 | |
| Number of N’s per 100 Kb | 0 | 0 | 0 | 0 | 0 | |
| Number of scaffolds >5000 bp | 29 | 11 | 28 | 53 | 23 | |
| Total length >5000 bp | 10,780,920 | 10,974,959 | 10,558,055 | 10,120,373 | 10,857,358 | |
| RagTag assembler | Assembly length (bp) | 11,092,545 | 11,019,016 | 10,925,846 | 10,870,339 | 11,021,815 |
| Number of scaffolds/chromosomes | 9 | 9 | 12 | 19 | 9 | |
| Number of N´s per 100 Kb | 3065 | 3.63 | 31.12 | 61.64 | 19.05 | |
| Number of scaffolds >5000 bp | 4 | 8 | 4 | 3 | 7 | |
| Total length >5000 bp | 11,025,073 | 11,000,234 | 10,766,609 | 10,606,285 | 10,966,127 | |
| Ploidy | haploid | haploid | haploid | haploid | haploid | |
| GC content (%) | 49.66 | 49.70 | 49.73 | 49.66 | 49.76 | |
| A1.18T | A1.5 | A1.7 | A1.19 | LB-NB-3.3 | |
|---|---|---|---|---|---|
| Protein coding genes | |||||
| Total number | 6092 | 6034 | 6067 | 6015 | 6038 |
| Range of protein lengths (aa) | 66–4974 | 63–4974 | 57–4974 | 60–4974 | 66–5293 |
| Average protein length (aa) | 557.6 | 556.6 | 550.9 | 543.5 | 518.3 |
| Non-coding RNAs | |||||
| microRNAs (miRNAs) | 32 | 32 | 33 | 31 | 21 |
| small RNAs (sRNA) | 20 | 21 | 22 | 20 | 23 |
| nuclear RNAs (snRNA) | 7 | 7 | 6 | 7 | 7 |
| nucleolar RNAs (snoRNA) | 93 | 91 | 99 | 94 | 98 |
| long noncoding RNAs (lncRNA) | 8 | 8 | 9 | 8 | 12 |
| ribosomal RNAs (rRNA) | 96 | 63 | 42 | 69 | 124 |
| transfer RNAs (tRNA) | 276 | 259 | 279 | 299 | 248 |
| Other | 29 | 32 | 32 | 35 | 32 |
| BUSCO Orthologs Ascomycota odb10 database | |||||
| Genome Completeness (%) | 93.5 | 94.4 | 93.4 | 90.7 | 93.6 |
| Complete BUSCOs | 1595 | 1611 | 1594 | 1547 | 1597 |
| Fragmented BUSCOs | 17 | 14 | 18 | 21 | 4 |
| Missing BUSCOs | 94 | 81 | 94 | 138 | 94 |
| Saccharomycetes odb10 database | |||||
| Genome Completeness (%) | 98.0 | 99.1 | 98.0 | 95.1 | 98.2 |
| Complete BUSCOs | 2094 | 2118 | 2094 | 2032 | 2099 |
| Fragmented BUSCOs | 14 | 11 | 12 | 17 | 13 |
| Missing BUSCOs | 29 | 8 | 31 | 88 | 25 |
| Eggnog-mapper functional annotation | |||||
| Genes with KO assigned | 3130 (51.4%) | 3129 (51.9%) | 3125 (51.6%) | 3130 (52.0%) | 3101 (51.4%) |
| Genes with COG assigned | 4180 (68.6%) | 4171 (69.1%) | 4166 (68.7%) | 4119 (68.5%) | 4141 (68.6%) |
| CAZymes functional annotation | |||||
| Number of genes annotated | 120 | 121 | 118 | 112 | 117 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Duarte, R.; Čadež, N.; Rito, T.; Drumonde-Neves, J.; Dominguez, Y.R.; Pais, C.; Sousa, M.J.; Soares, P. Whole-Genome Sequencing and Annotation of the Yeast Clavispora santaluciae Reveals Important Insights about Its Adaptation to the Vineyard Environment. J. Fungi 2022, 8, 52. https://doi.org/10.3390/jof8010052
Franco-Duarte R, Čadež N, Rito T, Drumonde-Neves J, Dominguez YR, Pais C, Sousa MJ, Soares P. Whole-Genome Sequencing and Annotation of the Yeast Clavispora santaluciae Reveals Important Insights about Its Adaptation to the Vineyard Environment. Journal of Fungi. 2022; 8(1):52. https://doi.org/10.3390/jof8010052
Chicago/Turabian StyleFranco-Duarte, Ricardo, Neža Čadež, Teresa Rito, João Drumonde-Neves, Yazmid Reyes Dominguez, Célia Pais, Maria João Sousa, and Pedro Soares. 2022. "Whole-Genome Sequencing and Annotation of the Yeast Clavispora santaluciae Reveals Important Insights about Its Adaptation to the Vineyard Environment" Journal of Fungi 8, no. 1: 52. https://doi.org/10.3390/jof8010052
APA StyleFranco-Duarte, R., Čadež, N., Rito, T., Drumonde-Neves, J., Dominguez, Y. R., Pais, C., Sousa, M. J., & Soares, P. (2022). Whole-Genome Sequencing and Annotation of the Yeast Clavispora santaluciae Reveals Important Insights about Its Adaptation to the Vineyard Environment. Journal of Fungi, 8(1), 52. https://doi.org/10.3390/jof8010052