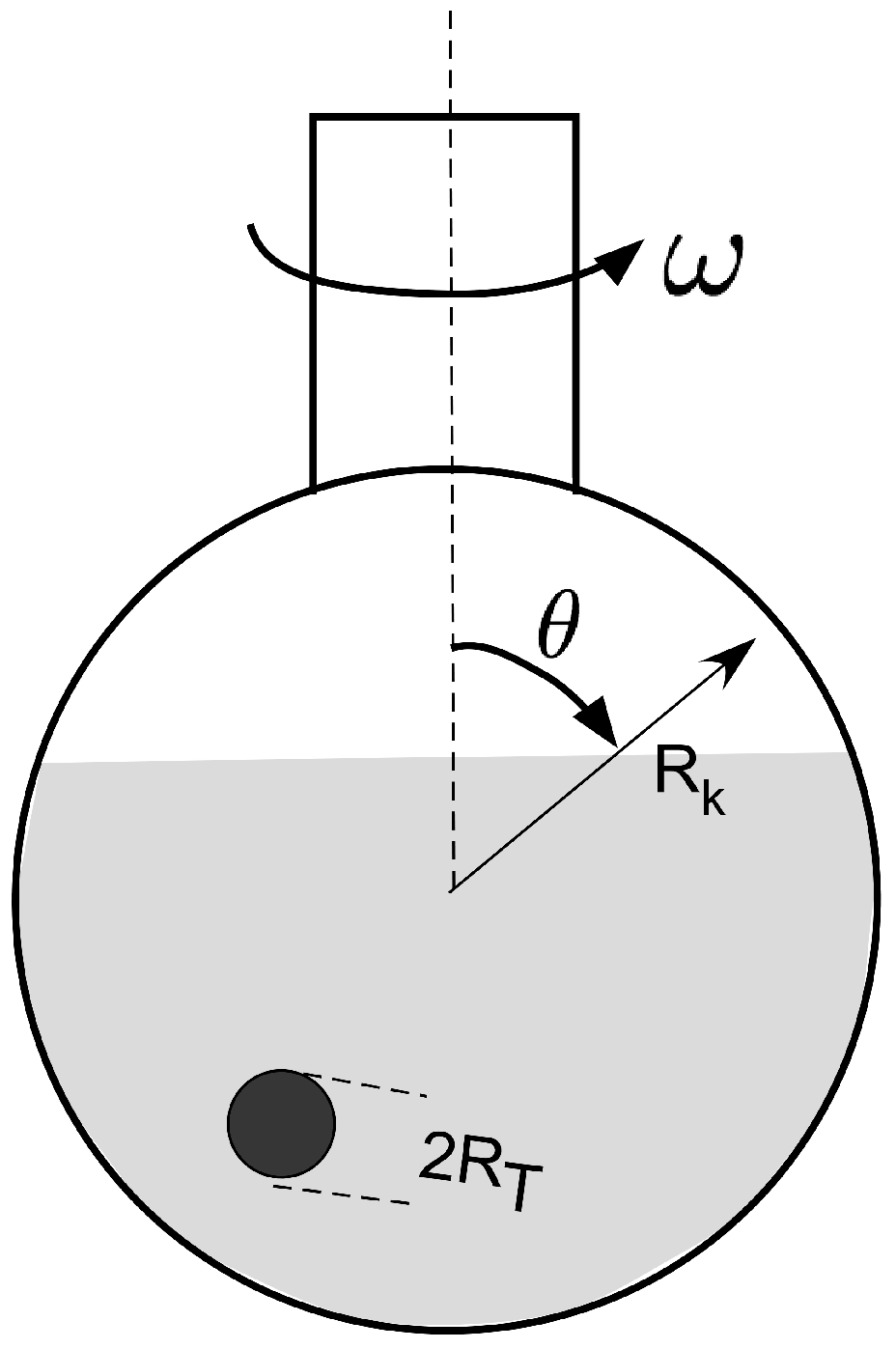
The data suggest that there is a non-linear dependence of gel time on droplet size, probably a logarithmic one. To the best of our knowledge such a relation is not reported in the literature. We therefore thought to develop an analytically tractable model reflecting specialties of our experimental set-up. We first have to take into account that the paraffin oil, which rotates in the bulb at low frequencies, fulfills a simple shear flow with an azimuthal velocity of
[
8], with
r as the radial coordinate in the bulb. The linear shear flow has a shear rate
. Let us assume that the drops are at an angle of 45
C in the oil. We then have
, with
f the rotation frequency. In the suspended drop, a shear flow also exists, which is according to [
9] described by a rotation period of
. The shear rate inside the drop is then simplified as
.
3.1. Aggregation by Shear Flow
In order to describe gelation, we simply take the Smoluchowski equation [
7], which describes aggregation like a kind of population balance: particles collide and aggregate forming bigger particles or clusters and particles vanish from a certain size class just due to this aggregation. If
denotes the number density of particles with volumes in the size class
the Smoluchowski equation reads
with the collision kernel
, which is essentially the total scattering cross section of two particles having a volume
v and
, multiplied with their relative velocity. The collision kernel
defines the way drops coagulate by e.g., Brownian motion or shear flows [
10,
11,
12]. We are not going to solve this equation for the given experimental conditions but will look for two issues which are easier to obtain then the full solution of the non-linear integro-differential Equation (
1). First, the number density of colloidal particles inside the solution (transforming to a gel)
can be obtained from integration of Equation (
1) over the particle volume yielding the relation
If there is no growth of the particles due to chemical reactions, then the volume fraction of particles denoted as Φ is constant
. The volume fraction and the average number density are always related as
via the average particle volume
. If there is no particle growth we have
. Since a shear flow exists in the suspended drop, the colloidal particles forming in it will also feel the shear, as mentioned above, and we simply assume that they thereby make collisions due to the linearly varying velocity inside the bulb. The collision volume for shear flows can be simplified as [
11]:
Inserting this into Equation (
2) and integrating leads to simple expression for the change in number density
This equation is easily solved yielding the average cluster volume and the number density of clusters.
It is important to notice that the average particle volume (aggregate volume) is unbounded. This is an essential feature of gelling systems [
3,
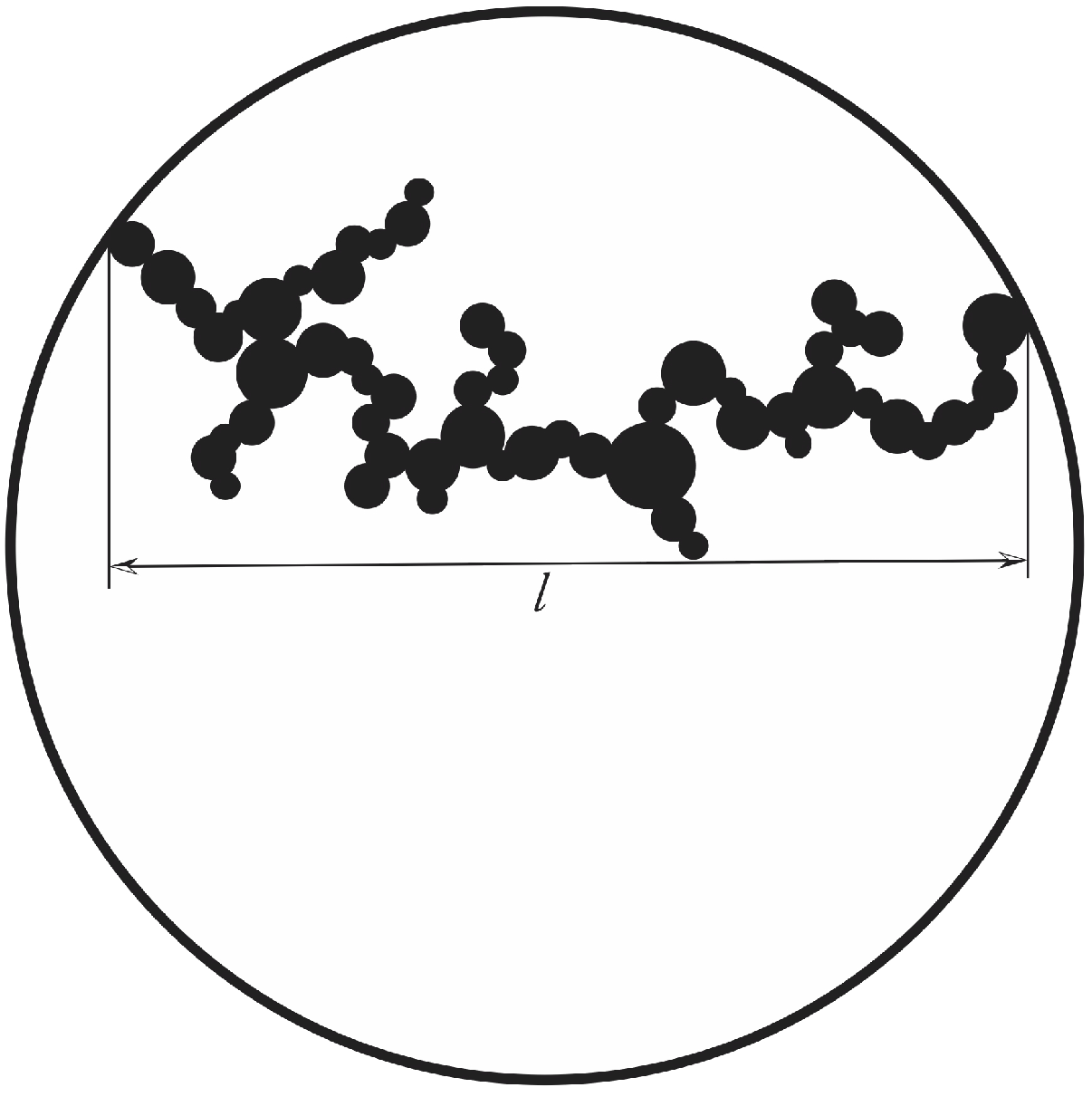
13]. Let us now define the gel time. This shall be done with the sketch shown in
Figure 4.
Figure 4.
Gelation is assumed to occur, if a spanning cluster of particles having all the same size expands from one side of the suspended drop to the other.
Figure 4.
Gelation is assumed to occur, if a spanning cluster of particles having all the same size expands from one side of the suspended drop to the other.
All primary particles shall have initially a volume
. They aggregate to clusters. Gelation occurs when these small particles make a spanning cluster of
. The number of particles in such a spanning cluster is
Since this number is identical to
, we obtain
This immediately gives the gel time as
This can be tested against the experimental values. Although a fit to the data (shown in
Figure 1) looks very well, it is misleading, since the shear rate in Equation (
9) also depends on drop volume. Inserting the above mentioned experimental power law relation, Equation (
30), yields an expression like
Such a relation cannot be fitted to the experimental results, since the gel time would depend almost linear on the drop volume, which is not the case. We nevertheless assume, that aggregation by shear collisions is an essential mechanism, since it gives a logarithmic dependence, but it is not sufficient to describe the experimental reality.
3.2. Concurrent Growth and Aggregation by Shear Flow
A better description of transformation from an RF solution to a gel should take into account, as mentioned in the introduction, that there is not only a size distribution of primary particles forming the network, but also a continuous growth of these, since there are abundant number of monomers, dimers
etc. that can react with given oligomers and particles or clusters even after the first onset of gelation (aging period of gels). We take this into account by modifying the Smoluchowski equation with a growth term [
12].
We again are not going to solve this equation for the size distribution, but first calculate the number density by integration of both sides of the equation with respect to the particle volume and then multiply both sides by the particle volume
v and integrate again over the particle volume. This yields the number density and the volume fraction of particle clusters as it varies with time. Performing the integration leads to two ordinary differential equations.
Instead of a constant volume fraction of RF-particles, we now have a time varying one
To evaluate these equations, we need an expression for the time varying change of particle volume
. For simplicity, we assume a constant volumetric growth of the particles with a chemical reaction of first order [
14] through its interface area
A at a rate
, meaning
Assuming that the particles remain spherical their surface area
A can be expressed in terms of particle volume and Equation (
13) be evaluated using
with
. Solving Equations (
12) and (
15) together leads first to an expression for the number density as a function of volume fraction.
Setting
as a new normalized variable of the volume fraction Φ with
yields
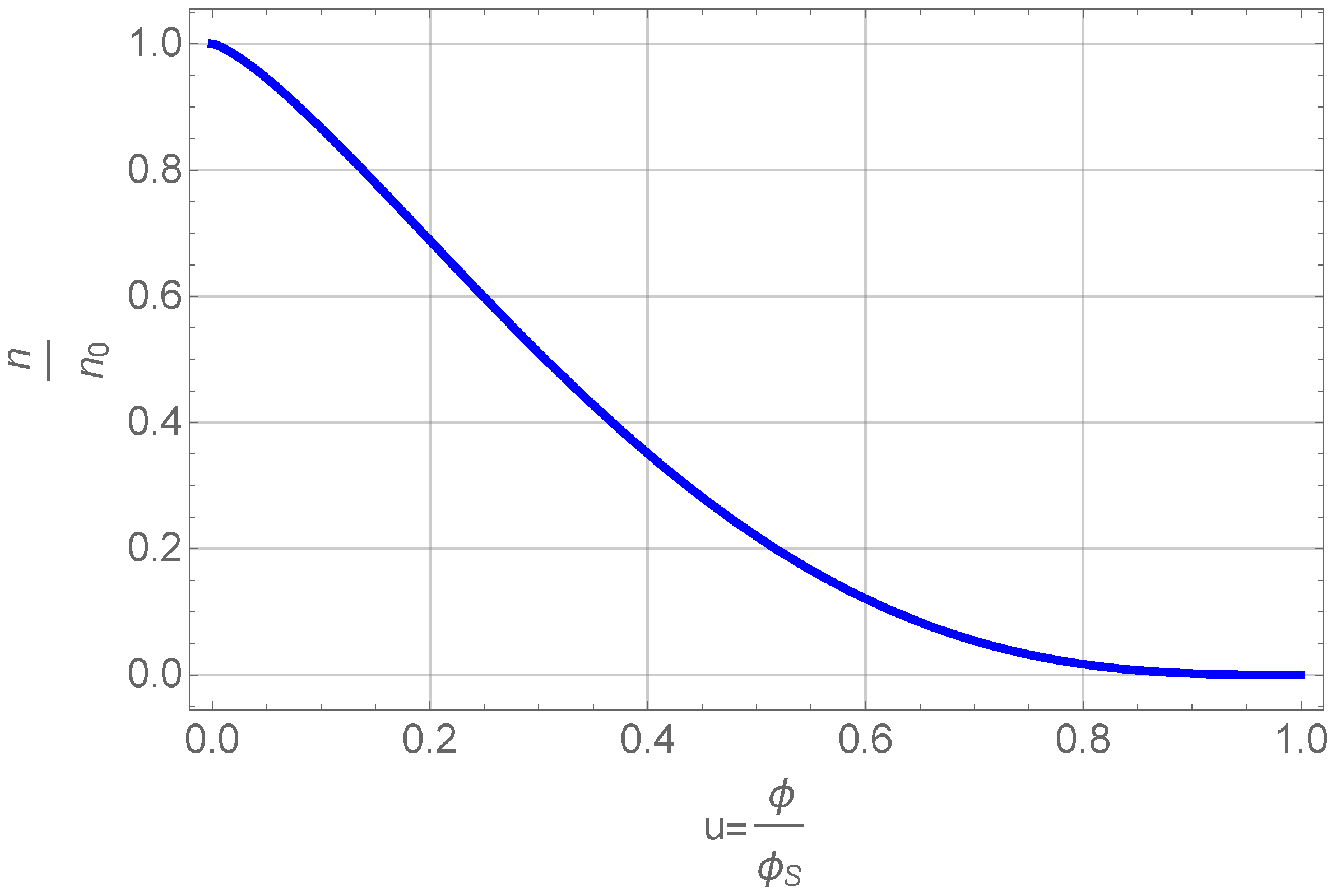
Figure 5.
Number density of clusters as a function of normalized volume fraction.
Figure 5.
Number density of clusters as a function of normalized volume fraction.
This figure shows that the number density of primary particles,
, that can grow and aggregate decreases with volume fraction. The volume fraction itself is a function of time (see below) and thus we get the simple result, that there is a finite time after which all primary particles are part of clusters or even one cluster. Inserting this Equation (
16) into Equation (
15) and using again
yields a differential equation for the volume fraction alone. Using again the normalized volume fraction
u and refining the time sale using
the differential equation for the normalized volume fraction reads
This equation can be integrated directly. The result is shown in
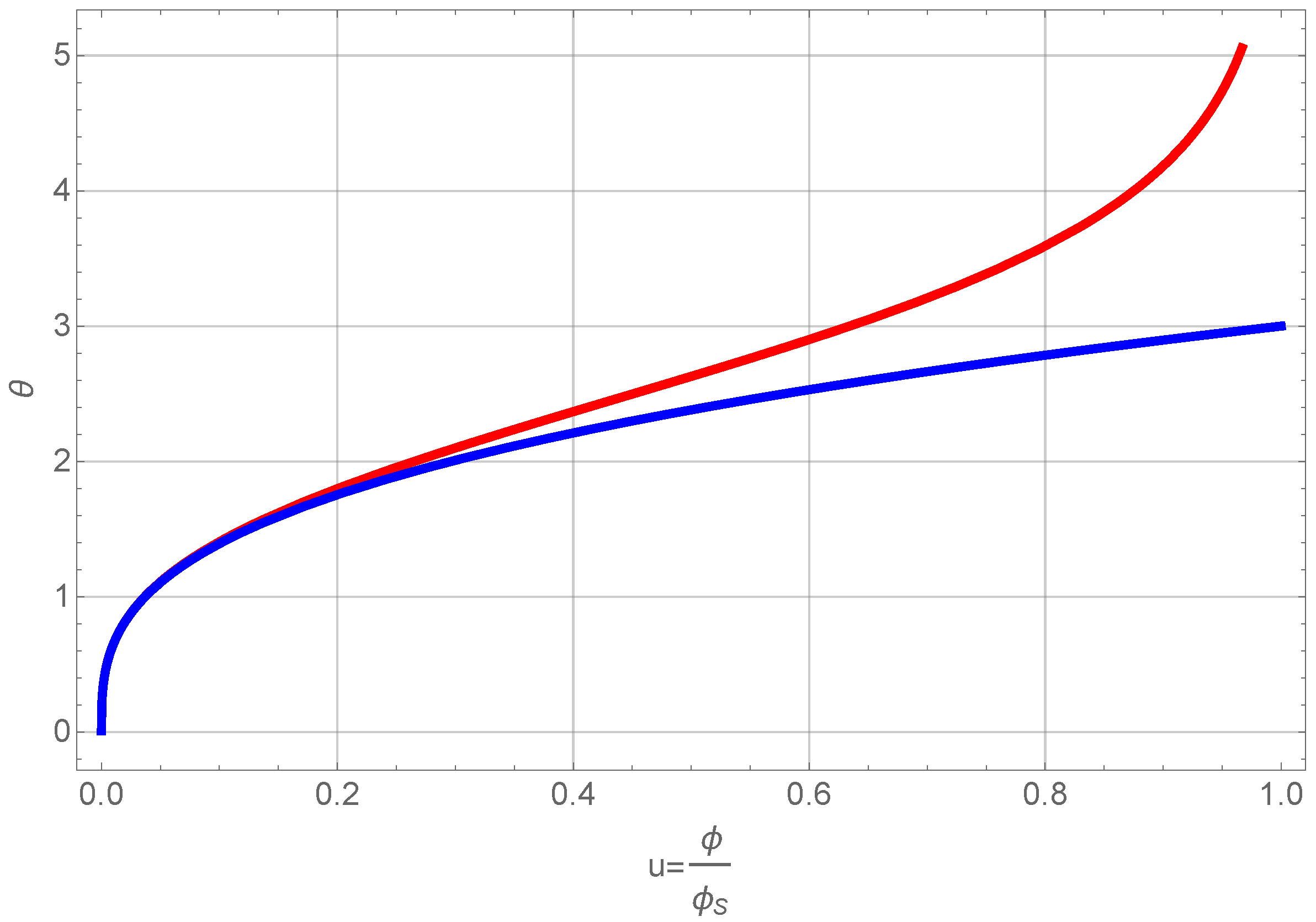
Figure 6.
Figure 6.
Normalized volume fraction of RF-particles as a function of scaled time
θ. The blue curve shows the full solution and the red one the approximate one of Equation (
21).
Figure 6.
Normalized volume fraction of RF-particles as a function of scaled time
θ. The blue curve shows the full solution and the red one the approximate one of Equation (
21).
We omit presenting the full solution of Equation (
20), but use only a series expansion at low volume fractions, since this allows us to further treat the problem analytically. A first order expansion of the normalized volume fraction as a function of normalized time gives
A comparison of the full solution and the approximate one is also presented in
Figure 6 showing that the approximation is valid for
. Integrating Equation (
12) directly and using the normalized time leads to an expression of the number density
and similarly one can derive for the average particle volume the relation
We now have to define an expression of the gelation time for aggregates consisting of particles with different sizes.
A sketch of such an aggregate is shown in
Figure 7. We can define that for a cluster of length
the number of particles at the gelation time
is given by
Figure 7.
If the particles for during aggregation the cluster consist of particles with different size. Still gelation is assumed to occur, if a spanning cluster of particles now having all the different sizes expands from one side of the suspended drop to the other.
Figure 7.
If the particles for during aggregation the cluster consist of particles with different size. Still gelation is assumed to occur, if a spanning cluster of particles now having all the different sizes expands from one side of the suspended drop to the other.
The number can be calculated from the number density
and the suspended drop volume
to be
. Putting these equations together leads to an expression for the gel time
Inserting the above expressions for the number density and the average particle volume into this expression using
, the approximate result given in Equation (
21) gives the relation (using normalized variables)
with
as the obvious definition of a scaled gelation time. Although this equation can be formally solved for
using the Lambert function, we do make a simplification which allows a direct comparison with the experimental data. Taking on both sides of Equation (
26), the logarithm allows one to give an expression for the gel time as
In this expression, we approximated the term stemming from the logarithmic form of Equation (
26), namely
being valid for normalized gel times between four and six. The result in Equation (
27) can be compared with the experimental results after transformation to non-normalized coordinates and using the power law relation between the shear rate and the drop volume. This gives the expression
with
numerical constants from the mentioned power law relation. To make a fit to the experimental data, we have to take into account, that the model assumes that a primary particle density
exists from the beginning. This, however, is in reality not the case. At the beginning of the experiment, the solution contains resorcinol, formaldehyde, water and the catalyst. The monomers have to react to build first dimers, trimers and oligomers. Before they can be treated as distinct primary particles, which are entrained in the shear flow field, it takes a certain incubation time
, as is well known for silica gels and discussed by Iler [
15] or in nucleation theory as discussed by Kelton and Greer [
16]. We therefore add to the above derived expression an incubation time and simplify it further by neglecting the expression in square brackets (an evaluation shows, that the logarithm is a very slowly varying function with drop volume and almost constant to around 2.8 for the drop volumes used and a primary particle density in the range of 10
to 10
and growth rates
in the range of 0.1 to 1 nm/s.). This leads us to our final equation
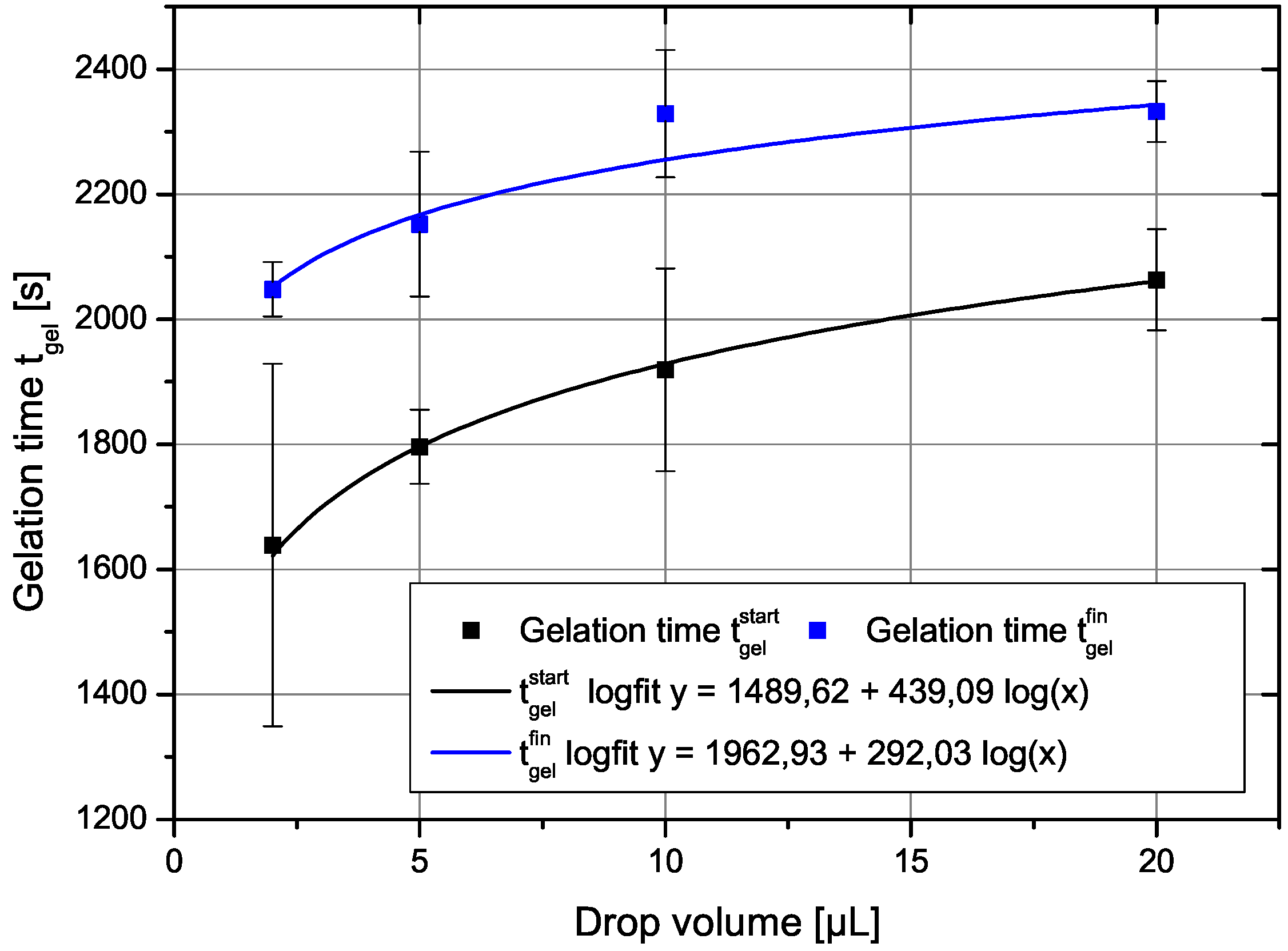
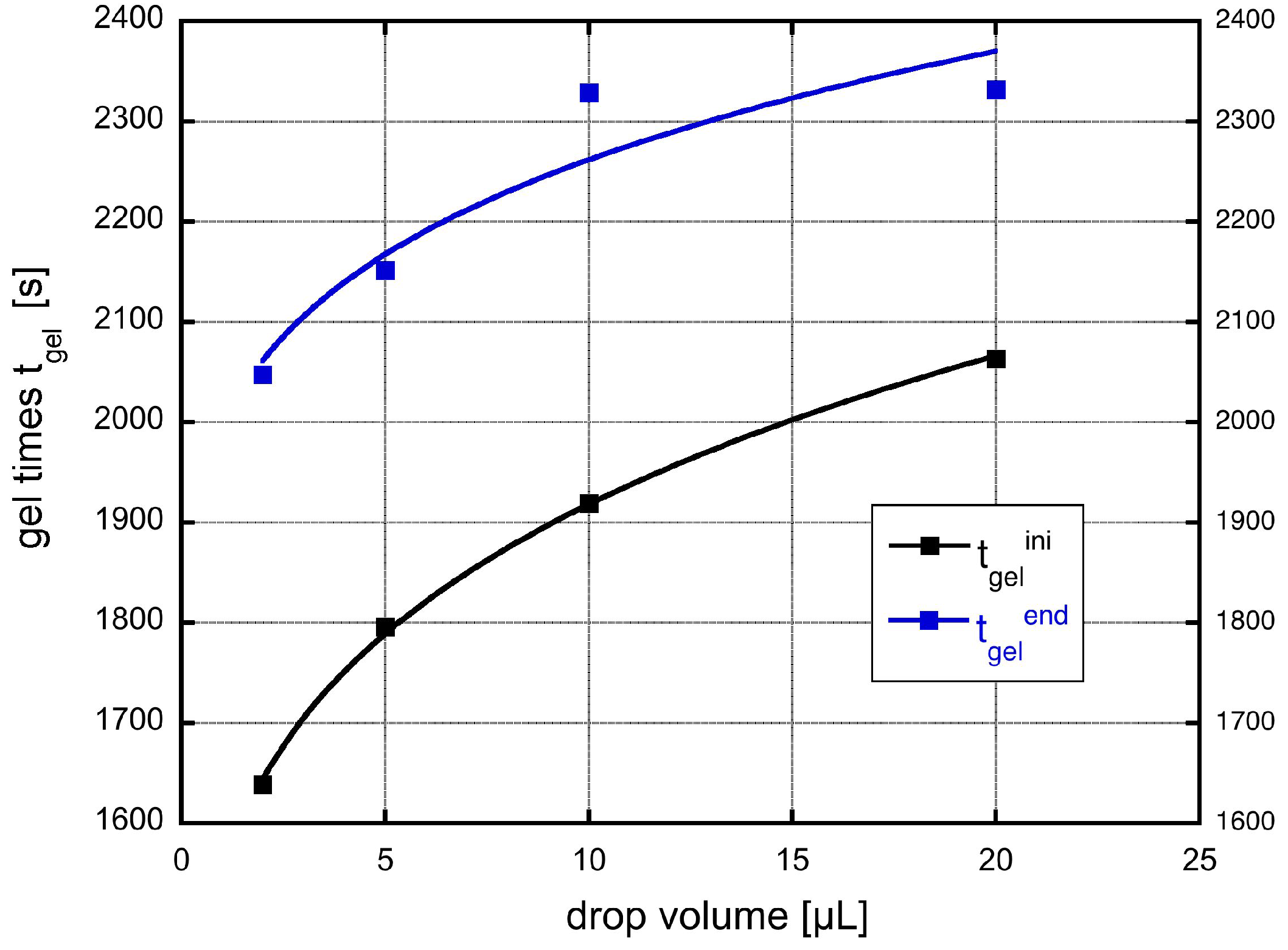
A fit of the experimental data to the theoretical expression is shown in
Figure 8. Here, we have used the original drop volumes given in μL.
We yield for the incubation times related to the onset of gelation and the final gelation point two different values,
and
s, respectively. The constant
c can be re-calculated to yield a value for the product
a value of 4
. This is for instance fulfilled for a reasonable initial concentration of primary particles of
m
and a growth rate
of 0.1 nm/s. Such a growth rate would mean that the particles grow in 1000 s to a size of 100 nm, which sounds reasonable and comparable to the SEM figures shown. The result also shows, that both effects are important, the growth, which leads in Equation (
28) to the dependence on shear rate to a power of
and the shear coagulation itself, reflected in the shear rate dependence. The microstructures shown in the SEM pictures of
Figure 3 also reflect the issue that, in the small droplets, the shear rate was higher than in the larger ones and, thus, they are more compact compared to those in which aggregation took place under mild shear conditions. To calculate the incubation time, another model for the chemical reactions kinetics would be needed.
Figure 8.
Measured gel-time compared with the theoretical expression given in Equation (
28).
Figure 8.
Measured gel-time compared with the theoretical expression given in Equation (
28).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
