Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications

Abstract
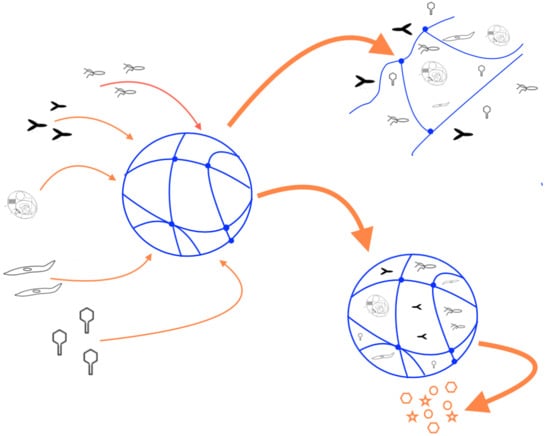
:
{kind=link}
{kind=link}
{kind=link}
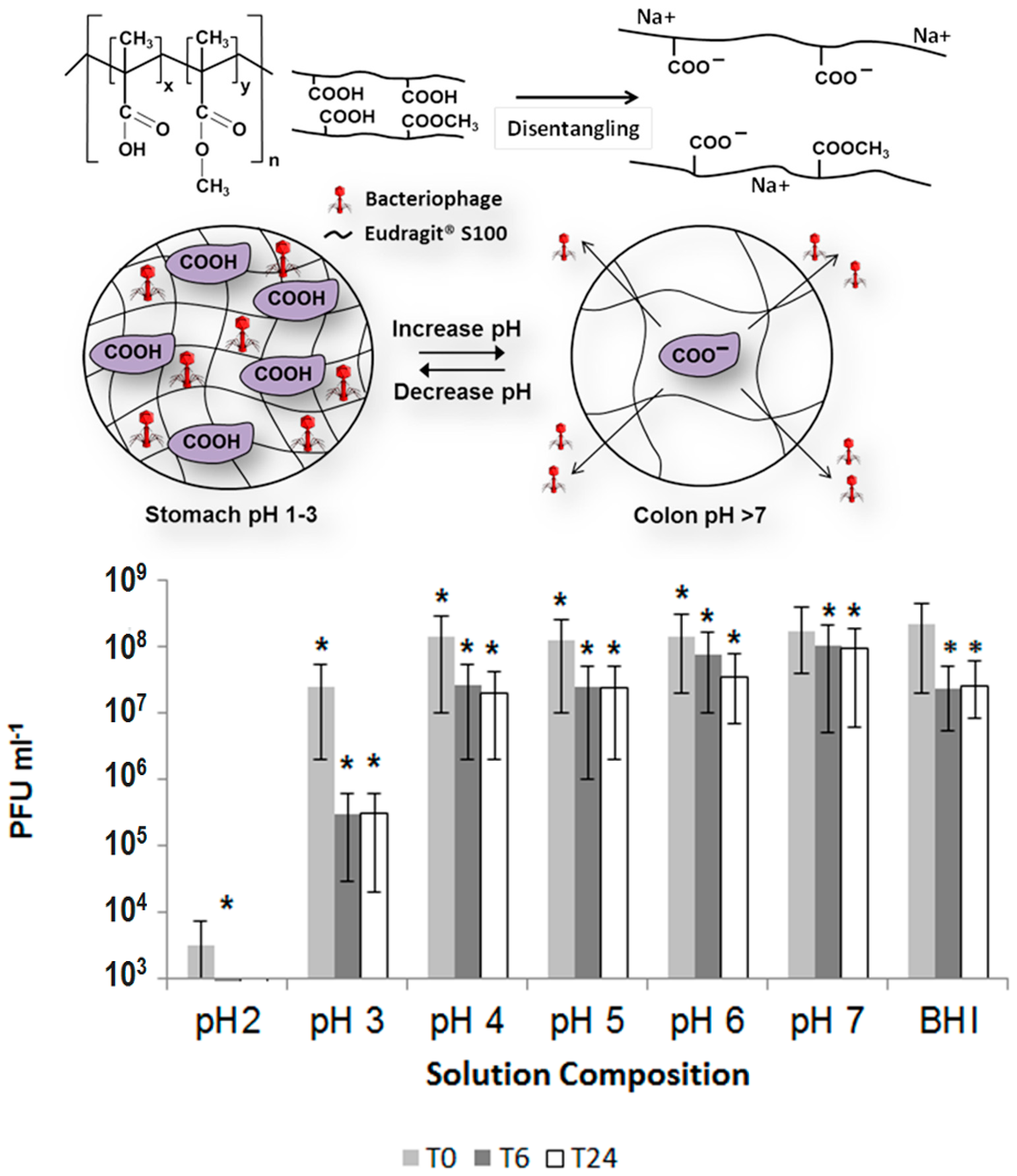{kind=link}
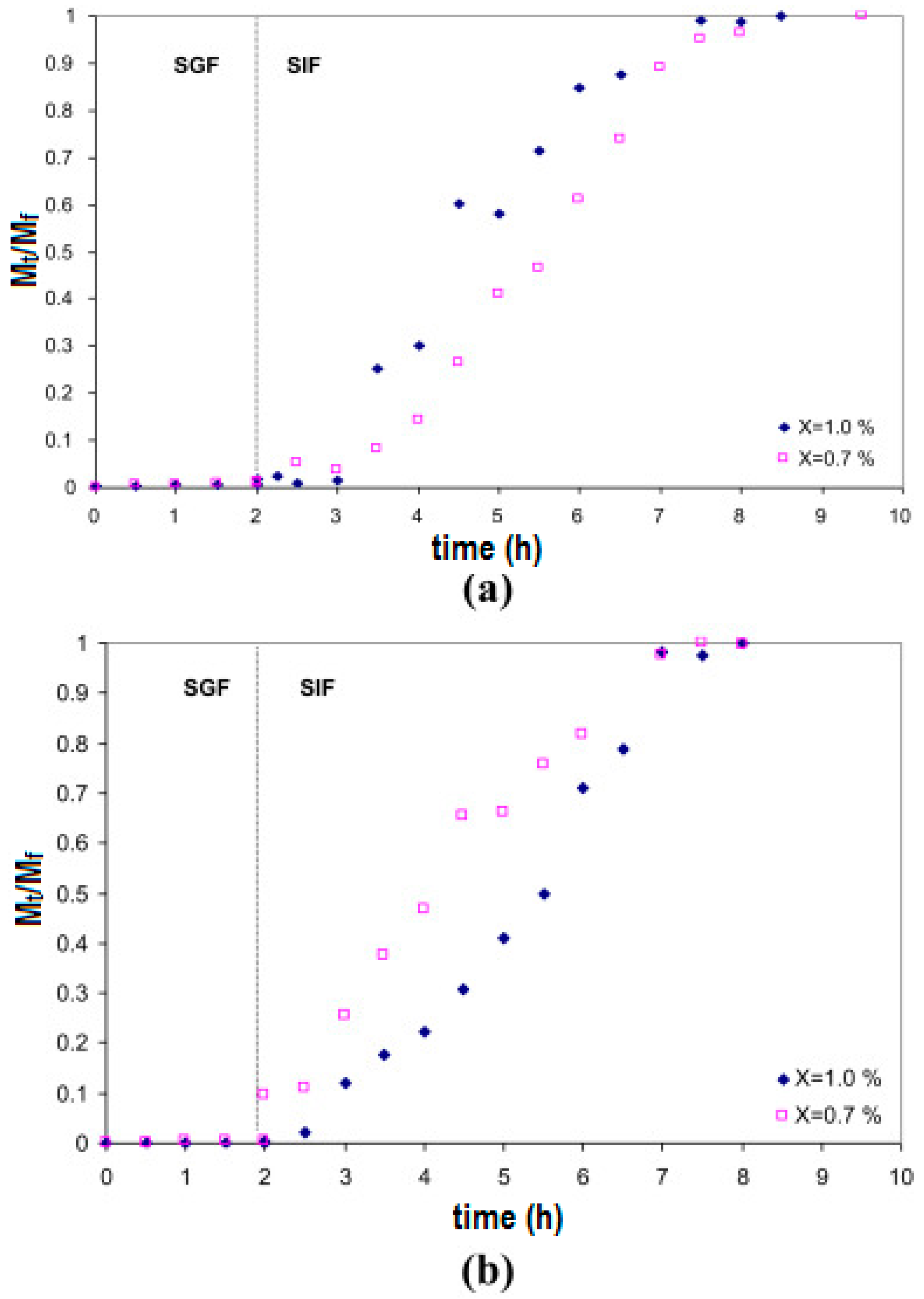{kind=link}
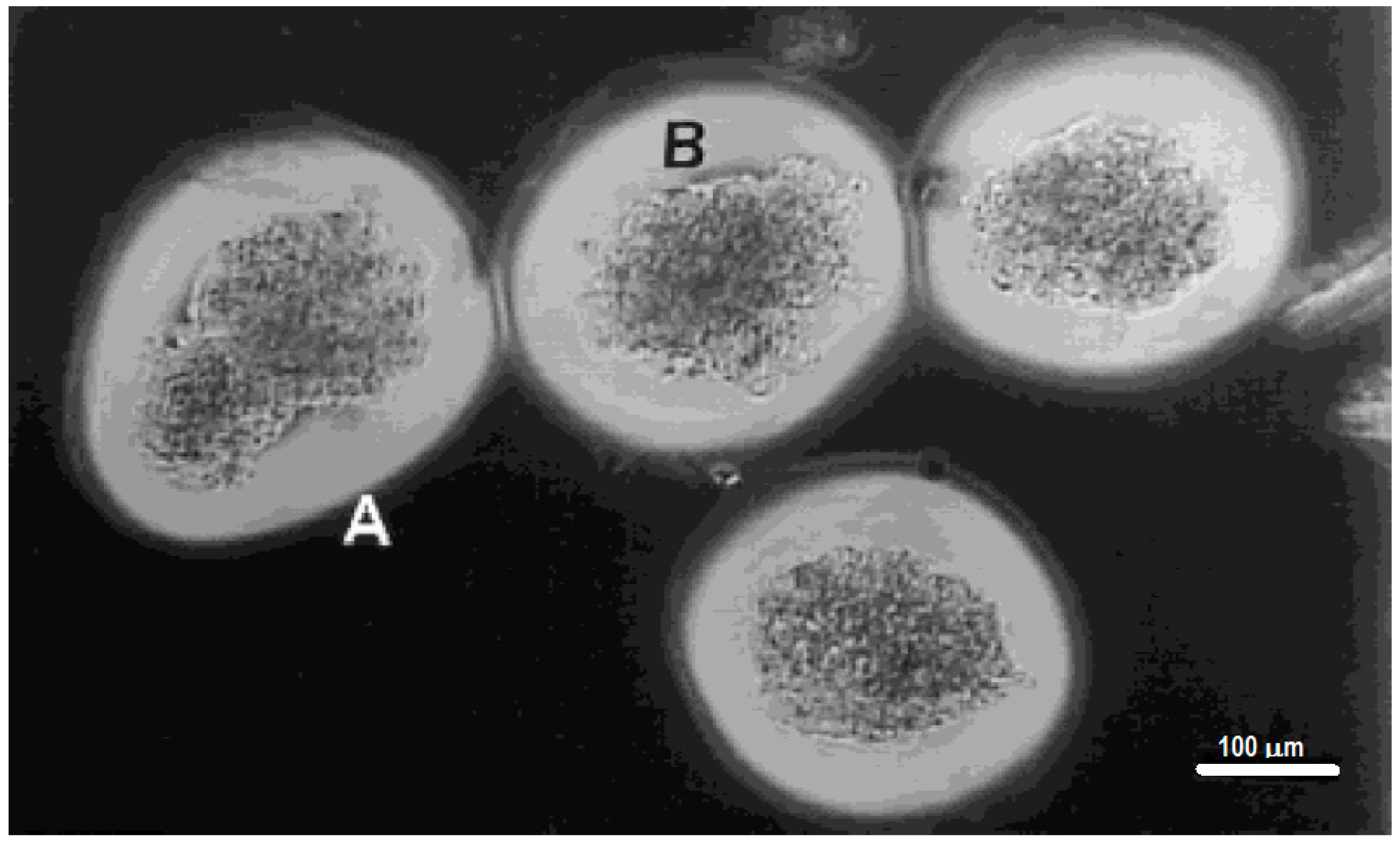{kind=link}
1. Introduction
2. Hydrogel Preparation
3. Encapsulation within Hydrogels for Medical Applications
4. Proteins and Biomolecules
Controlled Release of Encapsulated Molecules from Hydrogels
5. Viruses
6. Bacteria
7. Islet Cells
8. Tissue Engineering
9. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Ottenbrite, R.M.; Park, K.; Okano, T.; Peppas, N.A. Biomedical Applications of Hydrogels Handbook, 2010th ed.; Springer: New York, NY, USA, 2010; p. 432. [Google Scholar]
- Ratner, B.D.; Hoffman, A.S. Synthetic Hydrogels for Biomedical Applications. ACS Symp. Ser. 1976, 31, 1–36. [Google Scholar]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 13–36. [Google Scholar] [CrossRef]
- Song, G.; Zhang, L.; He, C.; Fang, D.-C.; Whitten, P.G.; Wang, H.; Jiang, L. Facile Fabrication of Tough Hydrogels Physically Cross-Linked by Strong Cooperative Hydrogen Bonding. Macromolecules 2013, 46, 7423–7435. [Google Scholar] [CrossRef]
- Zhang, G.; Lv, L.; Deng, Y.; Wang, C. Self-Healing Gelatin Hydrogels Cross-Linked by Combining Multiple Hydrogen Bonding and Ionic Coordination. Macromol. Rapid Commun. 2017, 38, 1700018. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhang, Y.; Li, Y.; Xua, B.; Liu, W. Hydrogen bonded and ionically crosslinked high strength hydrogels exhibiting Ca2+-triggered shape memory properties and volume shrinkage for cell detachment. J. Mater. Chem. B 2015, 3, 6347–6354. [Google Scholar] [CrossRef]
- Lee, A.L.Z.; Venkataraman, S.; Fox, C.H.; Coady, D.J.; Frank, C.W.; Hedrick, J.L.; Yang, Y.Y. Modular composite hydrogels from cholesterol-functionalized polycarbonates for antimicrobial applications. J. Mater. Chem. B 2015, 3, 6953–6963. [Google Scholar] [CrossRef]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A Thermoresponsive Hydrogel Based on Telechelic PEG End-Capped with Hydrophobic Dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A. Sustained release of human growth hormone from in situ forming hydrogels using self-assembly of fluoroalkyl-ended poly(ethylene glycol). Biomaterials 2005, 26, 5259–5266. [Google Scholar] [CrossRef] [PubMed]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A.; Johannsmann, D. Anomalous sorption in thin films of fluoroalkyl-ended poly(ethylene glycol)s. Langmuir 2002, 18, 8241–8245. [Google Scholar] [CrossRef]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A.; Johannsmann, D.; Hogen-Esch, T.E. Hydrogels with controlled, surface erosion characteristics from self-assembly of fluoroalkyl-ended poly(ethylene glycol). Macromolecules 2001, 34, 6409–6419. [Google Scholar] [CrossRef]
- Chang, X.; Geng, Y.; Cao, H.; Zhou, J.; Tian, Y.; Shan, G.; Bao, Y.; Wu, Z.L.; Pan, P. Dual-Crosslink Physical Hydrogels with High Toughness Based on Synergistic Hydrogen Bonding and Hydrophobic Interactions. Macromol. Rapid Commun. 2018, 1700806. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FA, USA, 2008. [Google Scholar]
- Hoffman, A.S. Hydrogels for Biomedical Applications. Adv. Drug. Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Maitra, J.; Shukla, V.K. Cross-linking in Hydrogels—A Review. Am. J. Polym. Sci. 2014, 4, 25–31. [Google Scholar]
- Cruise, G.M.; Scharp, D.S.; Hubbell, J.A. Characterization of permeability and network structure of interfacially photopolymerized poly(ethylene glycol) diacrylate hydrogels. Biomaterials 1998, 19, 1287–1294. [Google Scholar] [CrossRef]
- Cruise, G.M.; Hegre, O.D.; Scharp, D.S.; Hubbell, J.A. A sensitivity study of the key parameters in the interfacial photopolymerization of poly(ethylene glycol) diacrylate upon porcine islets. Biotechnol. Bioeng. 1998, 57, 655–665. [Google Scholar] [CrossRef]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Interfacial photopolymerization of poly(ethylene glycol)-based hydrogels upon alginate-poly(l-lysine) microcapsules for enhanced biocompatibility. Biomaterials 1993, 14, 1008–1016. [Google Scholar] [CrossRef]
- Hill-West, J.L.; Chowdhury, S.M.; Slepian, M.J.; Hubbell, J.A. Inhibition of thrombosis and intimal thickening by in situ photopolymerization of thin hydrogel barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5967–5971. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Shull, K.R.; Healy, K.E. Axisymmetric Adhesion Test To Examine the Interfacial Interactions between Biologically-Modified Networks and Models of the Extracellular Matrix. Langmuir 2003, 19, 1853–1860. [Google Scholar] [CrossRef]
- Healy, K.E.; Rezania, A.; Stile, R.A. Designing biomaterials to direct biological responses. Ann. N. Y. Acad. Sci. 1999, 875, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Sequential formation of hydrogel multilayers through surface initiated photopolymerization. Polym. Prepr. 2004, 45, 41–42. [Google Scholar]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F.A. Poly(ethylene glycol) diacrylate hydrogels on covalently attached eosin surface: Contact angle measurements of the surface. Polym. Prepr. 2003, 44, 206–207. [Google Scholar]
- Pathak, C.P.; Sawhney, A.S.; Hubbell, J.A. Rapid photopolymerization of immunoprotective gels in contact with cells and tissue. J. Am. Chem. Soc. 1992, 114, 8311–8312. [Google Scholar] [CrossRef]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Photopolymerization of Poly(Ethylene Glycol) Diacrylate on Eosin-Functionalized Surfaces. Langmuir 2004, 20, 8652–8658. [Google Scholar] [CrossRef] [PubMed]
- Papavasiliou, G.; Songprawat, P.; Perez-Luna, V.; Hammes, E.; Morris, M.; Chiu, Y.-C.; Brey, E. Three-Dimensional Patterning of Poly(Ethylene Glycol) Hydrogels Through Surface-Initiated Photopolymerization. Tissue Eng. Part C 2008, 14, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Kizilel, S.; Sawardecker, E.; Teymour, F.; Perez-Luna, V.H. Sequential formation of covalently bonded hydrogel multilayers through surface initiated photopolymerization. Biomaterials 2006, 27, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Odian, G. Principles of Polymerization, 4th ed.; Wiley-Interscience: New York, NY, USA, 2004. [Google Scholar]
- Moad, G.; Chiefari, J.; Chong, Y.K.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition—Fragmentation chain transfer (the life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Lowe, A.B.; Sumerlin, B.S.; Donovan, M.S.; McCormick, C.L. Facile Preparation of Transition Metal Nanoparticles Stabilized by Well-Defined (Co)polymers Synthesized via Aqueous Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2002, 124, 11562–11563. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Shimada, Y.; Mitsukami, Y.; Yamamoto, T.; Morishima, Y. pH-Responsive Micellization of Amphiphilic Diblock Copolymers Synthesized via Reversible Addition-Fragmentation Chain Transfer Polymerization. Macromolecules 2003, 36, 4208–4215. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rao, G.V.R.; Basame, S.B.; Keller, D.J.; Artyushkova, K.; Fulghum, J.E.; Lopez, G.P. Reversible Control of Free Energy and Topography of Nanostructured Surfaces. J. Am. Chem. Soc. 2004, 126, 8904–8905. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rao, G.V.R.; Ista, L.K.; Wu, Y.; Andrzejewski, B.P.; Sklar, L.A.; Ward, T.L.; Lopez, G.P. Control of molecular transport through stimuli-responsive ordered mesoporous materials. Adv. Mater. 2003, 15, 1262–1266. [Google Scholar] [CrossRef]
- Ma, H.; Hyun, J.; Stiller, P.; Chilkoti, A. “Non-Fouling” Oligo(ethylene glycol)-Functionalized Polymer Brushes Synthesized by Surface-Initiated Atom Transfer Radical Polymerization. Adv. Mater. 2004, 16, 338–341. [Google Scholar] [CrossRef]
- Zhao, B.; Brittain, W.J. Synthesis of Tethered Polystyrene-block-Poly(methyl methacrylate) Monolayer on a Silicate Substrate by Sequential Carbocationic Polymerization and Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 1999, 121, 3557–3558. [Google Scholar] [CrossRef]
- Kim, J.-B.; Bruening, M.L.; Baker, G.L. Surface-Initiated Atom Transfer Radical Polymerization on Gold at Ambient Temperature. J. Am. Chem. Soc. 2000, 122, 7616–7617. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Miller, P.J.; Shukla, N.; Immaraporn, B.; Gelman, A.; Luokala, B.B.; Siclovan, T.M.; Kickelbick, G.; Vallant, T.; Hoffmann, H.; et al. Polymers at Interfaces: Using Atom Transfer Radical Polymerization in the Controlled Growth of Homopolymers and Block Copolymers from Silicon Surfaces in the Absence of Untethered Sacrificial Initiator. Macromolecules 1999, 32, 8716–8724. [Google Scholar] [CrossRef]
- Li, X.; Husson, S.M. Adsorption of dansylated amino acids on molecularly imprinted surfaces: A surface plasmon resonance study. Biosens. Bioelectron. 2006, 22, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, Y.; Wang, X.; Liu, S. Fabrication of Hybrid Silica Nanoparticles Densely Grafted with Thermoresponsive Poly(N-isopropylacrylamide) Brushes of Controlled Thickness via Surface-Initiated Atom Transfer Radical Polymerization. Chem. Mater. 2008, 20, 101–109. [Google Scholar] [CrossRef]
- Chakraborty, S.; Bishnoi, S.W.; Pérez-Luna, V.H. Gold Nanoparticles with Poly(N-isopropyl acrylamide) Formed via Surface Initiated Atom Transfer Free Radical Polymerization Exhibit Unusually Slow Aggregation Kinetics. J. Phys. Chem. C 2010, 114, 5947–5955. [Google Scholar] [CrossRef]
- Lou, X.; Wang, C.; He, L. Core-Shell Au Nanoparticle Formation with DNA-Polymer Hybrid Coatings Using Aqueous ATRP. Biomacromolecules 2007, 8, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; He, L. Surface passivation using oligo(ethylene glycol) in ATRP-assisted DNA detection. Sens. Actuators B 2008, 129, 225–230. [Google Scholar] [CrossRef]
- Couet, J.; Biesalski, M. Surface-Initiated ATRP of N-Isopropylacrylamide from Initiator-Modified Self-Assembled Peptide Nanotubes. Macromolecules 2006, 39, 7258–7268. [Google Scholar] [CrossRef]
- Raula, J.; Shan, J.; Nuopponen, M.; Niskanen, A.; Jiang, H.; Kauppinen, E.I.; Tenhu, H. Synthesis of Gold Nanoparticles Grafted with a Thermoresponsive Polymer by Surface-Induced Reversible-Addition-Fragmentation Chain-Transfer Polymerization. Langmuir 2003, 19, 3499–3504. [Google Scholar] [CrossRef]
- Dimitrova, I.; Trzebickab, B.; Müllerc, A.H.E.; Dworakb, A.; Tsvetanov, C.B. Thermosensitive water-soluble copolymers with doubly responsive reversibly interacting entities. Prog. Polym. Sci. 2007, 32, 1275–1343. [Google Scholar] [CrossRef]
- He, M.; Jiang, H.Y.; Wang, R.; Xie, Y.; Zhao, W.F.; Zhao, C.S. A versatile approach towards multi-functional surfaces via covalently attaching hydrogel thin layers. J. Colloid Interface Sci. 2016, 484, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Rwei, S.P.; Anh, T.H.N.; Chiang, W.Y.; Way, T.F.; Hsu, Y.J. Synthesis and Drug Delivery Application of Thermo- and pH-Sensitive Hydrogels: Poly(β-CD-co-N-Isopropylacrylamide-co-IAM). Materials 2016, 9, 1003. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.L.; Liu, X.Y.; Xu, X.L.; Chen, S.; Zhu, X.Y.; Du, Y.C.; Li, F. Temperature and pH dual-responsive polyhedral oligomeric silsesquioxane/poly[2-(dimethyl amino)-ethyl methacrylate]-b-poly(N-isopropylacrylamide) hybrid materials synthesized via RAFT polymerization and thiol-ene reaction: Potential candidates as drug delivery systems. Mater. Chem. Phys. 2016, 179, 65–71. [Google Scholar]
- Li, S.; Zhao, Z.X.; Wu, W.; Ding, C.M.; Li, J.S. Dual pH-responsive micelles with both charge-conversional property and hydrophobic-hydrophilic transition for effective cellular uptake and intracellular drug release. Polym. Chem. 2016, 7, 2202–2208. [Google Scholar] [CrossRef]
- Zhu, H.Z.; You, L.Q.; Wei, H.L.; Wang, G.F.; Chu, H.J.; Zhu, J.; He, J. Preparation and Characterization of pH-Sensitive Hydrogel Microspheres Based on Atom Transfer Radical Polymerization. Polym. Eng. Sci. 2015, 55, 2775–2782. [Google Scholar] [CrossRef]
- Wang, H.Y.; Qin, A.W.; Li, X.; Zhao, X.Z.; Liu, D.P.; He, C.J. Biocompatible Amphiphilic Conetwork Based on Crosslinked Star Copolymers: A Potential Drug Carrier. J. Polym. Sci. Part A 2015, 53, 2537–2545. [Google Scholar] [CrossRef]
- Yang, B.G.; Wang, C.Y.; Zhang, Y.B.; Ye, L.; Qian, Y.F.; Shu, Y.; Wang, J.M.; Li, J.J.; Yao, F.L. A thermoresponsive poly(N-vinylcaprolactam-co-sulfobetaine methacrylate) zwitterionic hydrogel exhibiting switchable anti-biofouling and cytocompatibility. Polym. Chem. 2015, 6, 3431–3442. [Google Scholar] [CrossRef]
- Ghaemy, M.; Ziaei, S.; Alizadeh, R. Synthesis of pH-sensitive amphiphilic pentablock copolymers via combination of ring-opening and atom transfer radical polymerization for drug delivery. Eur. Polym. J. 2014, 58, 103–114. [Google Scholar] [CrossRef]
- Forbes, D.C.; Creixell, M.; Frizzell, H.; Peppas, N.A. Polycationic nanoparticles synthesized using ARGET ATRP for drug delivery. Eur. J. Pharm. Biopharm. 2013, 84, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Siegwart, D.J.; Lee, H.I.; Sherwood, G.; Peteanu, L.; Hollinger, J.O.; Kataoka, K.; Matyjaszewski, K. Biodegradable nanogels prepared by atom transfer radical polymerization as potential drug delivery carriers: Synthesis, biodegradation, in vitro release, and bioconjugation. J. Am. Chem. Soc. 2007, 129, 5939–5945. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, Y.Y.; Fan, X.D.; Huang, Y. Synthesis of thermo- and pH sensitive polymer beads containing beta-cyclodextrin and poly(N,N-dimethylaminoethyl metracrylate and their controlled drug release behavior. Acta Polym. Sin. 2005, 3, 357–362. [Google Scholar]
- Hoffman, A.S.; Cohn, D.; Hanson, S.R.; Harker, L.A.; Horbett, T.A.; Ratner, B.D.; Reynolds, L.O. Application of radiation-grafted hydrogels as blood-contacting biomaterials. Radiat. Phys. Chem. 1983, 22, 267–283. [Google Scholar] [CrossRef]
- Fittkau, M.H.; Zilla, P.; Bezuidenhout, D.; Lutolf, M.P.; Human, P.; Hubbell, J.A.; Davies, N. The selective modulation of endothelial cell mobility on RGD peptide containing surfaces by YIGSR peptides. Biomaterials 2005, 26, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Seliktar, D.; Zisch, A.H.; Lutolf, M.P.; Wrana, J.L.; Hubbell, J.A. MMP-2 sensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J. Biomed. Mater. Res. Part A 2004, 68, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Raeber, G.P.; Zisch, A.H.; Tirelli, N.; Hubbell, J.A. Cell-responsive synthetic hydrogels. Adv. Mater. 2003, 15, 888–892. [Google Scholar] [CrossRef]
- Lutolf, M.P.; Hubbell, J.A. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules 2003, 4, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.D.; Tirelli, N.; Hubbell, J.A. Photopolymerized hyaluronic acid-based hydrogels and interpenetrating networks. Biomaterials 2003, 24, 893–900. [Google Scholar] [CrossRef]
- Pratt Alison, B.; Weber Franz, E.; Schmoekel Hugo, G.; Muller, R.; Hubbell Jeffrey, A. Synthetic extracellular matrices for in situ tissue engineering. Biotechnol. Bioeng. 2004, 86, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halstenberg, S.; Panitch, A.; Rizzi, S.; Hall, H.; Hubbell Jeffrey, A. Biologically engineered protein-graft-poly(ethylene glycol) hydrogels: A cell adhesive and plasmin-degradable biosynthetic material for tissue repair. Biomacromolecules 2002, 3, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. Synthetic hydrogel niches that promote human mesenchymal stem cell viability. Matrix Biol. 2005, 24, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Benoit, D.S.W.; Anseth, K.S. The effect on osteoblast function of colocalized RGD and PHSRN epitopes on PEG surfaces. Biomaterials 2005, 26, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Anseth, K.S. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials 2002, 23, 4315–4323. [Google Scholar] [CrossRef]
- Kim, S.; Chung Eugene, H.; Gilbert, M.; Healy Kevin, E. Synthetic MMP-13 degradable ECMs based on poly(N-isopropylacrylamide-co-acrylic acid) semi-interpenetrating polymer networks. I. Degradation and cell migration. J. Biomed. Mater. Res. A 2005, 75, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Itle, L.J.; Koh, W.-G.; Pishko, M.V. Hepatocyte Viability and Protein Expression within Hydrogel Microstructures. Biotechnol. Prog. 2005, 21, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, S.C.; Ehrbar, M.; Halstenberg, S.; Raeber, G.P.; Schmoekel, H.G.; Hagenmueller, H.; Mueller, R.; Weber, F.E.; Hubbell, J.A. Recombinant Protein-co-PEG Networks as Cell-Adhesive and Proteolytically Degradable Hydrogel Matrixes. Part II: Biofunctional Characteristics. Biomacromolecules 2006, 7, 3019–3029. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, S.C.; Hubbell, J.A. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrixes. Part I: Development and physicochemical characteristics. Biomacromolecules 2005, 6, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Kopecek, J.; Yang, J. Hydrogels as smart biomaterials. Polym. Int. 2007, 56, 1078–1098. [Google Scholar] [CrossRef]
- Zhao, X.; Harris, J.M. Novel degradable poly(ethylene glycol) esters for drug delivery. ACS Symp. Ser. 1997, 680, 458–472. [Google Scholar]
- Zhao, X.; Harris, J.M. Novel degradable poly(ethylene glycol) hydrogels for controlled release of protein. J. Pharm. Sci. 1998, 87, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- West, J.L.; An, Y.; Hubbell, J.A. Photopolymerized intravascular hydrogels for protein delivery. In Proceedings of the International Symposium on Controlled Release of Bioactive Materials, Kyoto, Japan, 7–10 July 1996; Controlled Release Society: Kyoto, Japan, 1996; pp. 833–834. [Google Scholar]
- Sawhney, A.S.; Pathak, C.P.; van Rensburg, J.J.; Dunn, R.C.; Hubbell, J.A. Optimization of photopolymerized bioerodible hydrogel properties for adhesion prevention. J. Biomed. Mater. Res. 1994, 28, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, J.A.; Pathak, C.P.; Sawhney, A.S.; Desai, N.P.; Hill, J.L. Photopolymerizable Biodegradable Hydrogels as Tissue Contacting Material and Controlled-Release Carriers. U.S. Patent WO1993017669, 16 Septermber 1993. [Google Scholar]
- Reddy, S.K.; Anseth, K.S.; Bowman, C.N. Modeling of network degradation in mixed step-chain growth polymerizations. Polymer 2005, 46, 4212–4222. [Google Scholar] [CrossRef]
- Rice, M.A.; Anseth, K.S. Encapsulating chondrocytes in copolymer gels: Bimodal degradation kinetics influence cell phenotype and extracellular matrix development. J. Biomed. Mater. Res. Part A 2004, 70, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Bender, R.J.; Durand, K.L.; Anseth, K.S. Encapsulating chondrocytes in degrading PEG hydrogels with high modulus: Engineering gel structural changes to facilitate cartilaginous tissue production. Biotechnol. Bioeng. 2004, 86, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Durand, K.L.; Anseth, K.S. Manipulations in hydrogel chemistry control photoencapsulated chondrocyte behavior and their extracellular matrix production. J. Biomed. Mater. Res. Part A 2003, 67, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.J.; Bryant, S.J.; Anseth, K.S. Tailoring the Degradation of Hydrogels Formed from Multivinyl Poly(ethylene glycol) and Poly(vinyl alcohol) Macromers for Cartilage Tissue Engineering. Biomacromolecules 2003, 4, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Metters, A.T.; Bowman, C.N.; Anseth, K.S. Verification of scaling laws for degrading PLA-b-PEG-b-PLA hydrogels. AIChE J. 2001, 47, 1432–1437. [Google Scholar] [CrossRef]
- Mason, M.N.; Metters, A.T.; Bowman, C.N.; Anseth, K.S. Predicting Controlled-Release Behavior of Degradable PLA-b-PEG-b-PLA Hydrogels. Macromolecules 2001, 34, 4630–4635. [Google Scholar] [CrossRef]
- Lu, S.; Anseth, K.S. Release Behavior of High Molecular Weight Solutes from Poly(ethylene glycol)-Based Degradable Networks. Macromolecules 2000, 33, 2509–2515. [Google Scholar] [CrossRef]
- Anseth, K.S.; Metters, A.T.; Bryant, S.J.; Martens, P.J.; Elisseeff, J.H.; Bowman, C.N. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control. Release 2002, 78, 199–209. [Google Scholar] [CrossRef]
- Suggs, L.J.; Krishnan, R.S.; Garcia, C.A.; Peter, S.J.; Anderson, J.M.; Mikos, A.G. In vitro and in vivo degradation of poly(propylene fumarate-co-ethylene glycol) hydrogels. J. Biomed. Mater. Res. 1998, 42, 312–320. [Google Scholar] [CrossRef]
- Elvira, C.; Abraham, G.A.; Gallardo, A.; San Roman, J. Smart biodegradable hydrogels with applications in drug delivery and tissue engineering. In Biodegradable Systems in Tissue Engineering and Regenerative Medicine, 1st ed.; Reis, R.L., Román, J.S., Eds.; CRC Press: Boca Raton, FA, USA, 2005; pp. 493–508. [Google Scholar]
- Metters, A.T.; Anseth, K.S.; Bowman, C.N. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer 2000, 41, 3993–4004. [Google Scholar] [CrossRef]
- Murthy, N.; Thng, Y.X.; Schuck, S.; Xu, M.C.; Fréchet, J.M.J. A Novel Strategy for Encapsulation and Release of Proteins: Hydrogels and Microgels with Acid-Labile Acetal Cross-Linkers. J. Am. Chem. Soc. 2002, 124, 12398–12399. [Google Scholar] [CrossRef] [PubMed]
- Huebsch, N.; Gilbert, M.; Healy, K.E. Analysis of sterilization protocols for peptide-modified hydrogels. J. Biomed. Mater. Res. Part B 2005, 74, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Healy, K.E. Synthesis and Characterization of Injectable Poly(N-isopropylacrylamide-co-acrylic acid) Hydrogels with Proteolytically Degradable Cross-Links. Biomacromolecules 2003, 4, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Raeber, G.P.; Lutolf, M.P.; Hubbell, J.A. Molecularly engineered PEG hydrogels: A novel model system for proteolytically mediated cell migration. Biophys. J. 2005, 89, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Lauer-Fields, J.L.; Schmoekel, H.G.; Metters, A.T.; Weber, F.E.; Fields, G.B.; Hubbell, J.A. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. USA 2003, 100, 5413–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeloew, C.; Segura, T.; Hubbell, J.A.; Frey, P. The effect of enzymatically degradable poly(ethylene glycol) hydrogels on smooth muscle cell phenotype. Biomaterials 2007, 29, 314–326. [Google Scholar] [CrossRef] [PubMed]
- West, J.L.; Hubbell, J.A. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules 1999, 32, 241–244. [Google Scholar] [CrossRef]
- Goetsch, K.P.; Bracher, M.; Bezuidenhout, D.; Zilla, P.; Davies, N.H. Regulation of tissue ingrowth into proteolytically degradable hydrogels. Acta Biomater. 2015, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Miller, J.S.; Moon, J.J.; West, J.L. Proteolytically degradable hydrogels with a fluorogenic substrate for studies of cellular proteolytic activity and migration. Biotechnol. Prog. 2005, 21, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Dikovsky, D.; Bianco-Peled, H.; Seliktar, D. Proteolytically Degradable Photo-Polymerized Hydrogels Made From PEG–Fibrinogen Adducts. Adv. Eng. Mater. 2010, 12, B200–B209. [Google Scholar] [CrossRef]
- Wade, R.J.; Bassin, E.J.; Rodell, C.B.; Burdick, J.A. Protease-degradable electrospun fibrous hydrogels. Nat. Commun. 2015, 6, 6639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutolf, M.P.; Weber, F.E.; Schmoekel, H.G.; Schense, J.C.; Kohler, T.; Mueller, R.; Hubbell, J.A. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat. Biotechnol. 2003, 21, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Elisseeff, J.; McIntosh, W.; Fu, K.; Blunk, T.; Langer, R. Controlled-release of IGF-I and TGF-b1 in a photopolymerizing hydrogel for cartilage tissue engineering. J. Orthop. Res. 2001, 19, 1098–1104. [Google Scholar] [CrossRef]
- Goessl, A.; Tirelli, N.; Hubbell, J.A. A hydrogel system for stimulus-responsive, oxygen-sensitive in situ gelation. J. Biomater. Sci. Polym. Ed. 2004, 15, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lutolf, M.P.; Hubbell, J.A.; Hunziker, E.B.; Wong, M. Bovine Primary Chondrocyte Culture in Synthetic Matrix Metalloproteinase-Sensitive Poly(ethylene glycol)-Based Hydrogels as a Scaffold for Cartilage Repair. Tissue Eng. 2004, 10, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Hill-West, J.L.; Chowdhury, S.M.; Sawhney, A.S.; Pathak, C.P.; Dunn, R.C.; Hubbell, J.A. Prevention of postoperative adhesions in the rat by in situ photopolymerization of bioresorbable hydrogel barriers. Obstet. Gynecol. 1994, 83, 59–64. [Google Scholar] [PubMed]
- Suggs, L.J.; Mikos, A.G. Development of poly(propylene fumarate-co-ethylene glycol) as an injectable carrier for endothelial cells. Cell Transplant. 1999, 8, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Burghardt, W.R.; Healy, K.E. Synthesis and Characterization of Injectable Poly(N-isopropylacrylamide)-Based Hydrogels That Support Tissue Formation in Vitro. Macromolecules 1999, 32, 7370–7379. [Google Scholar] [CrossRef]
- Ruel-Gariepy, E.; Leroux, J.C. In situ-forming hydrogels—Review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef] [PubMed]
- Nih, L.R.; Carmichael, S.T.; Segura, T. Hydrogels for brain repair after stroke: An emerging treatment option. Curr. Opin. Biotechnol. 2016, 40, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Modification of islet of Langerhans surfaces with immunoprotective poly(ethylene glycol) coatings via interfacial photopolymerization. Biotechnol. Bioeng. 1994, 44, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Cruise, G.M.; Hegre, O.D.; Lamberti, F.V.; Hager, S.R.; Hill, R.; Scharp, D.S.; Hubbell, J.A. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly(ethylene glycol) diacrylate membranes. Cell Transplant. 1999, 8, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Pathak, C.P.; Sawhney, A.S.; Hubbell, J.A. In situ photopolymerization and gelation of water-soluble monomers: A new approach for local administration of peptide drugs. Polym. Prepr. 1992, 33, 65–66. [Google Scholar]
- Hubbell, J.A.; Pathak, C.P.; Sawhney, A.S. In vivo photopolymerization of PEG-based biodegradable hydrogels for the control of wound healing. Polym. Prepr. 1993, 34, 846–847. [Google Scholar]
- Rice, M.A.; Martens, P.J.; Bryant, S.J.; Mahoney, M.J.; Bowman, C.N.; Anseth, K.S. Photopolymerization of synthetic hydrogel niches for 3D cell culture and tissue regeneration. Polym. Prepr. 2004, 45, 11–12. [Google Scholar]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Bioerodible Hydrogels Based on Photopolymerized Poly(Ethylene Glycol)-co-Poly(Alpha-Hydroxy Acid) Diacrylate Macromers. Macromolecules 1993, 26, 581–587. [Google Scholar] [CrossRef]
- Cellesi, F.; Weber, W.; Fussenegger, M.; Hubbell, J.A.; Tirelli, N. Towards a fully synthetic substitute of alginate: Optimization of a thermal gelation/chemical cross-linking scheme (“tandem” gelation) for the production of beads and liquid-core capsules. Biotechnol. Bioeng. 2004, 88, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Cellesi, F.; Tirelli, N.; Hubbell, J.A. Towards a fully-synthetic substitute of alginate: Development of a new process using thermal gelation and chemical cross-linking. Biomaterials 2004, 25, 5115–5124. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Healy, K.E. Poly(N-isopropylacrylamide)-Based Semi-interpenetrating Polymer Networks for Tissue Engineering Applications. 1. Effects of Linear Poly(acrylic acid) Chains on Phase Behavior. Biomacromolecules 2002, 3, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Healy, K.E. Thermo-Responsive Peptide-Modified Hydrogels for Tissue Regeneration. Biomacromolecules 2001, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Chung, E.; Burghardt, W.R.; Healy, K.E. Poly(N-isopropylacrylamide)-based semi-interpenetrating polymer networks for tissue engineering applications. Effects of linear poly(acrylic acid) chains on rheology. J. Biomater. Sci. Polym. Ed. 2004, 15, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Turturro, S.B.; Guthrie, M.J.; Appel, A.A.; Drapala, P.W.; Brey, E.M.; Pérez-Luna, V.H.; Mieler, W.F.; Kang-Mieler, J.J. The effects of cross-linked thermo-responsive PNIPAAm-based hydrogel injection on retinal function. Biomaterials 2011, 32, 3620–3626. [Google Scholar] [CrossRef] [PubMed]
- Drapala, P.W.; Brey, E.M.; Mieler, W.F.; Venerus, D.C.; Derwent, J.J.K.; Pérez-Luna, V.H. Role of Thermo-responsiveness and Poly(ethylene glycol) Diacrylate Cross-link Density on Protein Release from Poly(N-isopropylacrylamide) Hydrogels. J. Biomater. Sci. Polym. Ed. 2011, 22, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Drapala, P.W.; Jiang, B.; Chiu, Y.-C.; Mieler, W.F.; Brey, E.M.; Kang-Mieler, J.J.; Pérez-Luna, V.H. The Effect of Glutathione as Chain Transfer Agent in PNIPAAm-Based Thermo-responsive Hydrogels for Controlled Release of Proteins. Pharm. Res. 2014, 31, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Sheth, S.; Dunn, A.; Zustiak, S.P.; Sell, S.A. Sustained release of multicomponent platelet-rich plasma proteins from hydrolytically degradable PEG hydrogels. J. Biomed. Mater. Res. A 2017, 105, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Mostafalu, P.; Kiaee, G.; Giatsidis, G.; Khalilpour, A.; Nabavinia, M.; Dokmeci, M.R.; Sonkusale, S.; Orgill, D.P.; Tamayol, A.; Khademhosseini, A. A Textile Dressing for Temporal and Dosage Controlled Drug Delivery. Adv. Funct. Mater. 2017, 27, 1702399. [Google Scholar] [CrossRef]
- Kasiewicz, L.N.; Whitehead, K.A. Recent advances in biomaterials for the treatment of diabetic foot ulcers. Biomater. Sci. 2017, 5, 1962–1975. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Lan, Y.; Zuo, Q.; Li, C.; Zhang, Y.; Guo, R.; Xue, W. Acceleration of skin regeneration in full-thickness burns by incorporation of bFGF-loaded alginate microspheres into a CMCS-PVA hydrogel. J. Tissue Eng. Regener. Med. 2017, 11, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Chen, D.; Shen, C.; Shen, J.; Zhao, H.; He, Y. Platelet-Rich Plasma-Loaded Poly(d,l-lactide)-Poly(ethylene glycol)-Poly(d,l-lactide) Hydrogel Dressing Promotes Full-Thickness Skin Wound Healing in a Rodent Model. Int. J. Mol. Sci. 2016, 17, 1001. [Google Scholar] [CrossRef] [PubMed]
- Hajimiri, M.; Shahverdi, S.; Esfandiari, M.A.; Larijani, B.; Atyabi, F.; Rajabiani, A.; Dehpour, A.R.; Amini, M.; Dinarvand, R. Preparation of hydrogel embedded polymer-growth factor conjugated nanoparticles as a diabetic wound dressing. Drug Dev. Ind. Pharm. 2016, 42, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Takei, T.; Nakahara, H.; Tanaka, S.; Nishimata, H.; Yoshida, M.; Kawakami, K. Effect of chitosan-gluconic acid conjugate/poly(vinyl alcohol) cryogels as wound dressing on partial-thickness wounds in diabetic rats. J. Mater. Sci. Mater. Med. 2013, 24, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Moura, L.I.F.; Dias, A.M.A.; Carvalho, E.; de Sousa, H.C. Recent advances on the development of wound dressings for diabetic foot ulcer treatment—A review. Acta Biomater. 2013, 9, 7093–7114. [Google Scholar] [CrossRef] [PubMed]
- Fabiilli, M.L.; Wilson, C.G.; Padilla, F.; Martin-Saavedra, F.M.; Fowlkes, J.B.; Franceschi, R.T. Acoustic droplet-hydrogel composites for spatial and temporal control of growth factor delivery and scaffold stiffness. Acta Biomater. 2013, 9, 7399–7409. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.P.; Morgado, P.I.; Miguel, S.P.; Coutinho, P.; Correia, I.J. Dextran-based hydrogel containing chitosan microparticles loaded with growth factors to be used in wound healing. Mater. Sci. Eng. C 2013, 33, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.; Morck, D.; Schultz, C. Treatment of corneal defects with delayed re-epithelization with a medical device/drug delivery system for epidermal growth factor. Clin. Exp. Ophthalmol. 2012, 40, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Mishra, D.; Das, T.; Maiti, T.K. Wound pH-Responsive Sustained Release of Therapeutics from a Poly(NIPAAm-co-AAc) Hydrogel. J. Biomater. Sci. Polym. Ed. 2012, 23, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.W.; Byun, J.H.; Oh, S.H.; Kim, T.H.; Park, J.S.; Rho, G.J.; Lee, J.H. Multivalent ion-based in situ gelling polysaccharide hydrogel as an injectable bone graft. Carbohydr. Polym. 2018, 180, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Coletta, D.J.; Ibanez-Fonseca, A.; Missana, L.R.; Jammal, M.V.; Vitelli, E.J.; Aimone, M.; Zabalza, F.; Issa, J.P.M.; Alonso, M.; Rodriguez-Cabello, J.C.; et al. Bone Regeneration Mediated by a Bioactive and Biodegradable Extracellular Matrix-Like Hydrogel Based on Elastin-Like Recombinamers. Tissue Eng. Part A 2017, 23, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Jordan, J.; Roy, A.; Gottardi, R.; Fedorchak, M.V.; Kumta, P.N.; Little, S.R. Programmed Platelet-Derived Growth Factor-BB and Bone Morphogenetic Protein-2 Delivery from a Hybrid Calcium Phosphate/Alginate Scaffold. Tissue Eng. Part A 2017, 23, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Santovena, A.; Monzon, C.; Delgado, A.; Evora, C.; Llabres, M.; Farina, J.B. Development of a standard method for in vitro evaluation of Triamcinolone and BMP-2 diffusion mechanism from thermosensitive and biocompatible composite hyaluronic acid-pluronic hydrogels. J. Drug Deliv. Sci. Technol. 2017, 42, 284–291. [Google Scholar] [CrossRef]
- Seo, B.B.; Koh, J.T.; Song, S.C. Tuning physical properties and BMP-2 release rates of injectable hydrogel systems for an optimal bone regeneration effect. Biomaterials 2017, 122, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Wang, J.L.; Wu, J.J.; Zhang, J.; Wan, Y.; Wu, H. Injectable hydrogels embedded with alginate microspheres for controlled delivery of bone morphogenetic protein-2. Biomed. Mater. 2016, 11, 025010. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.M.R.; Black, C.R.M.; Dawson, J.I.; Oreffo, R.O.C. A review of hydrogel use in fracture healing and bone regeneration. J. Tissue Eng. Regener. Med. 2016, 10, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Priddy, L.B.; Esancy, C.; Li, M.T.A.; Stevens, H.Y.; Jiang, X.; Tran, L.; Rowe, D.W.; Guldberg, R.E. Hydrogel-based Delivery of rhBMP-2 Improves Healing of Large Bone Defects Compared With Autograft. Clin. Orthop. Relat. Res. 2015, 473, 2885–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelbach, R.J.; Fransen, P.; Pulido, D.; D’Este, M.; Duttenhoefer, F.; Sauerbier, S.; Freiman, T.; Niemeyer, P.; Albericio, F.; Alini, M.; et al. Injectable Hyaluronan Hydrogels with Peptide-Binding Dendrimers Modulate the Controlled Release of BMP-2 and TGF-beta 1. Macromol. Biosci. 2015, 15, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Brockmeyer, P.; Kramer, K.; Krohn, S.; Kauffmann, P.; Mauth, C.; Dard, M.; Schliephake, H.; Gruber, R.M. Influence of synthetic polyethylene glycol hydrogels on new bone formation during mandibular augmentation procedures in Goettingen minipigs. J. Mater. Sci. Mater. Med. 2015, 26, 194. [Google Scholar] [CrossRef] [PubMed]
- Metzger, S.; Lienemann, P.S.; Ghayor, C.; Weber, W.; Martin, I.; Weber, F.E.; Ehrbar, M. Modular Poly(ethylene glycol) Matrices for the Controlled 3D-Localized Osteogenic Differentiation of Mesenchymal Stem Cells. Adv. Healthc. Mater. 2015, 4, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Song, C.C.; Du, F.S.; Li, Z.C. Supersensitive Oxidation-Responsive Biodegradable PEG Hydrogels for Glucose-Triggered Insulin Delivery. ACS Appl. Mater. Interfaces 2017, 9, 25905–25914. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.L.; Wei, W.; Li, J.J.; Zuo, G.C.; Pan, X.H.; Su, T.; Zhang, J.F.; Dong, W. Salecan-Based pH-Sensitive Hydrogels for Insulin Delivery. Mol. Pharm. 2017, 14, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, G.H.; Yu, W.J.; Liu, D.P.; Chen, H.; Liu, Y.K.; Huang, Q.; Tong, Z.Z.; Yao, J.M.; Kong, X.D. A composite hydrogel system containing glucose-responsive nanocarriers for oral delivery of insulin. Mater. Sci. Eng. C 2016, 69, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, X.Y.; Zhang, Y.; Shang, Q. Fabrication and evaluation of a novel polymeric hydrogel of carboxymethyl chitosan-g-polyacrylic acid (CMC-g-PAA) for oral insulin delivery. RSC Adv. 2016, 6, 52858–52867. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Mukherjee, D.; Mishra, R.; Kundu, P.P. Development of pH sensitive polyurethane-alginate nanoparticles for safe and efficient oral insulin delivery in animal models. RSC Adv. 2016, 6, 41835–41846. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Chakraborty, S.; Bhattacharya, S.; Mishra, R.; Kundu, P.P. pH-sensitive chitosan/alginate core-shell nanoparticles for efficient and safe oral insulin delivery. Int. J. Biol. Macromol. 2015, 72, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Cao, Y.; Song, X.F.; Zhang, Z.; Zhuang, X.L.; He, C.L.; Chen, X.S. Biodegradable, pH-Responsive Carboxymethyl Cellulose/Poly(Acrylic Acid) Hydrogels for Oral Insulin Delivery. Macromol. Biosci. 2014, 14, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, A.; Al-Remawi, M.; Maghrabi, I.; Hamaidi, M.; Jaber, N. Development of insulin loaded mesoporous silica injectable particles layered by chitosan as a controlled release delivery system. Int. J. Pharm. 2014, 461, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Dang, T.T.; Ma, M.L.; Tang, B.C.; Cheng, H.; Jiang, S.; Dong, Y.Z.; Zhang, Y.L.; Anderson, D.G. Glucose-Responsive Microgels Integrated with Enzyme Nanocapsules for Closed-Loop Insulin Delivery. ACS Nano 2013, 7, 6758–6766. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Sarkar, K.; Soam, S.; Kundu, P.P. Formulation of pH-responsive carboxymethyl chitosan and alginate beads for the oral delivery of insulin. J. Appl. Polym. Sci. 2013, 129, 835–845. [Google Scholar] [CrossRef]
- Liu, G.; Ma, R.J.; Ren, J.; Li, Z.; Zhang, H.X.; Zhang, Z.K.; An, Y.L.; Shi, L.Q. A glucose-responsive complex polymeric micelle enabling repeated on-off release and insulin protection. Soft Matter 2013, 9, 1636–1644. [Google Scholar] [CrossRef]
- Deat-Laine, E.; Hoffart, V.; Cardot, J.M.; Subirade, M.; Beyssac, E. Development and in vitro characterization of insulin loaded whey protein and alginate microparticles. Int. J. Pharm. 2012, 439, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mundargi, R.C.; Rangaswamy, V.; Aminabhavi, T.M. pH-Sensitive oral insulin delivery systems using Eudragit microspheres. Drug Dev. Ind. Pharm. 2011, 37, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ma, G.H.; Wang, L.Y.; Wu, J.; Su, Z.G. Hollow quaternized chitosan microspheres increase the therapeutic effect of orally administered insulin. Acta Biomater. 2010, 6, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Builders, P.F.; Kunle, O.O.; Okpaku, L.C.; Builders, M.O.; Attama, A.A.; Adikwu, M.U. Preparation and evaluation of mucinated sodium alginate microparticles for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2008, 70, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, A.; Pruthi, V. Oral insulin delivery by polymeric nanospheres. J. Biomed. Nanotechnol. 2007, 3, 68–74. [Google Scholar] [CrossRef]
- Sajeesh, S.; Sharma, C.P. Interpolymer complex microparticles based on polymethacrylic acid-chitosan for oral insulin delivery. J. Appl. Polym. Sci. 2006, 99, 506–512. [Google Scholar]
- Sajeesh, S.; Sharma, C.P. Poly methacrylic acid-alginate semi-IPN microparticles for oral delivery of insulin: A preliminary investigation. J. Biomater. Appl. 2004, 19, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.K.K.; Sharma, C.P. Oral insulin—A perspective. J. Biomater. Appl. 2003, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Victor, S.P.; Sharma, C.P. Stimuli sensitive polymethacrylic acid microparticles (PMAA)—Oral insulin delivery. J. Biomater. Appl. 2002, 17, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Wu, Y.D.; Ye, H.B.; Yu, S.J.; He, C.L.; Chen, X.S. Interleukin-15 and cisplatin co-encapsulated thermosensitive polypeptide hydrogels for combined immuno-chemotherapy. J. Control. Release 2017, 255, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Bobbala, S.; Tamboli, V.; McDowell, A.; Mitra, A.K.; Hook, S. Novel Injectable Pentablock Copolymer Based Thermoresponsive Hydrogels for Sustained Release Vaccines. AAPS J. 2016, 18, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Walke, S.; Srivastava, G.; Nikalje, M.; Doshi, J.; Kumar, R.; Ravetkar, S.; Doshi, P. Fabrication of chitosan microspheres using vanillin/TPP dual crosslinkers for protein antigens encapsulation. Carbohydr. Polym. 2015, 128, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.H.; Li, S.; Howard, O.M.Z.; Dunlap, M.; Trivett, A.; Schneider, J.P.; Oppenheim, J.J. Enhanced immunostimulatory effects of DNA-encapsulated peptide hydrogels. Biomaterials 2015, 53, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Hariyadi, D.M.; Ma, Y.; Wang, Y.; Bostrom, T.; Malouf, J.; Turner, M.S.; Bhandari, B.; Coombes, A.G.A. The potential for production of freeze-dried oral vaccines using alginate hydrogel microspheres as protein carriers. J. Drug Deliv. Sci. Technol. 2014, 24, 178–184. [Google Scholar] [CrossRef]
- Yun, Y.H.; Goetz, D.J.; Yellen, P.; Chen, W.L. Hyaluronan microspheres for sustained gene delivery and site-specific targeting. Biomaterials 2004, 25, 147–157. [Google Scholar] [CrossRef]
- Murthy, N.; Xu, M.C.; Schuck, S.; Kunisawa, J.; Shastri, N.; Frechet, J.M.J. A macromolecular delivery vehicle for protein-based vaccines: Acid-degradable protein-loaded microgels. Proc. Natl. Acad. Sci. USA 2003, 100, 4995–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowersock, T.L.; HogenEsch, H.; Torregrosa, S.; Borie, D.; Wang, B.; Park, H.; Park, K. Induction of pulmonary immunity in cattle by oral administration of ovalbumin in alginate microspheres. Immunol. Lett. 1998, 60, 37–43. [Google Scholar] [CrossRef]
- Fletcher, N.A.; Babcock, L.R.; Murray, E.A.; Krebs, M.D. Controlled delivery of antibodies from injectable hydrogels. Mater. Sci. Eng. C 2016, 59, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Blasi, L.; Argentiere, S.; Morello, G.; Palama, I.; Barbarella, G.; Cingolani, R.; Gigli, G. Uptake and distribution of labeled antibodies into pH-sensitive microgels. Acta Biomater. 2010, 6, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Lira, J.C.; Camacho-Frias, E.; Pena-Alvarez, A.; Vera-Avila, L.E. Preparation and characterization of a sol-gel immunosorbent doped with 2,4-D antibodies. Chem. Mater. 2003, 15, 154–161. [Google Scholar] [CrossRef]
- Markowitz, M.A.; Turner, D.C.; Martin, B.D.; Gaber, B.P. Diffusion and transfer of antibody proteins from a sugar-based hydrogel. Appl. Biochem. Biotechnol. 1997, 68, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in form of slabs, sphere, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Zarzycki, R.; Modrzejewska, Z.; Nawrotek, K. Drug release from hydrogel matrices. Ecol. Chem. Eng. S 2010, 17, 117–136. [Google Scholar]
- Cohen, M.H.; Turnbull, D. Molecular transport in liquids and glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef]
- Masaro, L.; Zhu, X.X. Physical models of diffusion for polymer solutions, gels and solids. Prog. Polym. Sci. 1999, 24, 731–775. [Google Scholar] [CrossRef]
- Bird, R.B.; Stewart, W.E.; Lightfoot, E.N. Transport Phenomena; John Wiley and Sons: Toronto, ON, Canada, 1960. [Google Scholar]
- Mackie, J.S.; Meares, P. The diffusion of electrolytes in a cation-exchange resin membrane I. Theoretical. Proc. R. Soc. A 1955, 232, 498–509. [Google Scholar] [CrossRef]
- Amsden, B. Solute Diffusion within Hydrogels. Mechanisms and Models. Macromolecules 1998, 31, 8382–8395. [Google Scholar] [CrossRef]
- Amsden, B. An Obstruction-Scaling Model for Diffusion in Homogenous Hydrogels. Macromolecules 1999, 32, 874–879. [Google Scholar] [CrossRef]
- Lustig, S.R.; Peppas, N.A. Solute Diffusion in Swollen Membranes. IX. Scaling Laws for Solute Diffusion in Gels. J. Appl. Polym. Sci. 1988, 36, 735–747. [Google Scholar] [CrossRef]
- Cukier, R.I. Diffusion of Brownian spheres in semidilute polymer solutions. Macromolecules 1984, 17, 252–255. [Google Scholar] [CrossRef]
- Phillips, R.J.; Deen, W.M.; Brady, J.F. Hindered transport of spherical macromolecules in fibrous membranes and gels. AIChE J. 1989, 35, 1761–1769. [Google Scholar] [CrossRef]
- Jackson, G.W.; James, D.F. The permeability of fibrous porous media. Can. J. Chem. Eng. 1986, 64, 364–374. [Google Scholar] [CrossRef]
- Ogston, A.G. The spaces in a uniform random suspension of fibres. Trans. Faraday Soc. 1958, 54, 1754–1757. [Google Scholar] [CrossRef]
- Dini, C.; Islan, G.A.; Urraza, P.J.D.; Castro, G.R. Novel Biopolymer Matrices for Microencapsulation of Phages: Enhanced Protection Against Acidity and Protease Activity. Macromol. Biosci. 2012, 12, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Dini, C.; Islan, G.A.; Castro, G.R. Characterization and Stability Analysis of Biopolymeric Matrices Designed for Phage-Controlled Release. Appl. Biochem. Biotechnol. 2014, 174, 2031–2047. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Taylor, C.M. What’s new in haemolytic uraemic syndrome? Eur. J. Pediatr. 2008, 167, 965. [Google Scholar] [CrossRef] [PubMed]
- Dini, C.; Urraza, P.J.D. Isolation and selection of coliphages as potential biocontrol agents of enterohemorrhagic and Shiga toxin-producing E. coli (EHEC and STEC) in cattle. J. Appl. Microbiol. 2010, 109, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.E.; Lamont, J.T. Clostridium difficile infection: A worldwide disease. Gut Liver 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E.S. Consequences of Clostridium difficile infection: Understanding the healthcare burden. Clin. Microbiol. Infect. 2012, 18, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Polyzos, K.; Patouni, K.; Rafailidis, P.I.; Samonis, G.; Falagas, M.E. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: A systematic review of the evidence. Int. J. Antimicrob. Agents 2012, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Vladisavljević, G.T.; Clokie, M.R.J.; Malik, D.J. Microencapsulation of Clostridium difficile specific bacteriophages using microfluidic glass capillary devices for colon delivery using pH triggered release. PLoS ONE 2017, 12, e0186239. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.M.; Stickler, D.J.; Mobley, H.L.T.; Shirtliff, M.E. Complicated catheter-associated urinary tract infections due to Escherichia coli and Proteus mirabilis. Clin. Microbiol. Rev. 2008, 21, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Stickler, D.J.; Morgan, S.D. Modulation of crystalline Proteus mirabilis biofilm development on urinary catheters. J. Med. Microbiol. 2006, 55, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.; Mobley, H. Merging mythology and morphology: The multifaceted lifestyle of Proteus mirabilis. Nat. Rev. Microbiol. 2012, 10, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Milo, S.; Hathaway, H.; Nzakizwanayo, J.; Alves, D.R.; Esteban, P.P.; Jonesbe, B.V.; Jenkins, A.T.A. Prevention of encrustation and blockage of urinary catheters by Proteus mirabilis via pH-triggered release of bacteriophage. J. Mater. Chem. B 2017, 5, 5403–5411. [Google Scholar] [CrossRef]
- Skoumal, M.; Seidlits, S.; Shin, S.; Shea, L. Localized Lentivirus Delivery Via Peptide Interactions. Biotechnol. Bioeng. 2016, 113, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.J.; Retamales, J.; Palza, H.; Bastías, R. Encapsulation of specific Salmonella Enteritidis phage f3αSE on alginate-spheres as a method for protection and dosification. Electron. J. Biotechnol. 2018, 31, 57–60. [Google Scholar] [CrossRef]
- Bean, J.E.; Alves, D.R.; Laabei, M.; Esteban, P.P.; Thet, N.T.; Enright, M.C.; Jenkins, A.T.A. Triggered Release of Bacteriophage K from Agarose/Hyaluronan Hydrogel Matrixes by Staphylococcus aureus Virulence Factors. Chem. Mater. 2014, 26, 7201–7208. [Google Scholar] [CrossRef] [Green Version]
- Fuller, R. Probiotics in man and animals. J. Appl. Bacteriol. 1989, 66, 365–378. [Google Scholar] [PubMed]
- Hood, S.K.; Zottola, E.A. Effect of low pH on the ability of Lactobacillus acidophilus to survive and adhere to human intestinal-cells. J. Food Sci. 1988, 53, 1514–1516. [Google Scholar] [CrossRef]
- Hughes, D.B.; Hoover, D.G. Bifidobacteria: Their potential for use in American dairy products. Food Technol. 1991, 45, 74–83. [Google Scholar]
- Shah, N.P.; Lankaputhra, W.E.V. Improving viability of Lactobacillus acidophilus and Bifidobacterium spp. in yogurt. Int. Dairy J. 1997, 7, 349–356. [Google Scholar] [CrossRef]
- Argin, S.; Kofinas, P.; Lo, Y.M. The cell release kinetics and the swelling behavior of physically crosslinked xanthanechitosan hydrogels in simulated gastrointestinal conditions. Food Hydrocoll. 2014, 40, 138–144. [Google Scholar] [CrossRef]
- Le Lay, C.; Dridi, L.; Bergeron, M.G.; Ouellette, M.; Fliss, I.L. Nisin is an effective inhibitor of Clostridium difficile vegetative cells and spore germination. J. Med. Microbiol. 2016, 65, 169–175. [Google Scholar] [PubMed]
- Xu, M.; Gagné-Bourque, F.; Dumont, M.-J.; Jabaji, S. Encapsulation of Lactobacillus casei ATCC 393 cells and evaluation of their survival after freeze-drying, storage and under gastrointestinal conditions. J. Food Eng. 2016, 168, 52–59. [Google Scholar] [CrossRef]
- Yeung, T.W.; Üçok, E.F.; Tiani, K.A.; McClements, D.J.; Sela, D.A. Microencapsulation in Alginate and Chitosan Microgels to Enhance Viability of Bifidobacterium longum for Oral Delivery. Front. Microbiol. 2016, 7, 494. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Zhang, Z. Bioencapsulation by compression coating of probiotic bacteria for their protection in an acidic medium. Process Biochem. 2005, 40, 3346–3351. [Google Scholar] [CrossRef]
- Strand, B.L.; Coron, A.E.; Skjak-Braek, G. Current and Future Perspectives on Alginate Encapsulated Pancreatic Islet. STEM CELLS Transl. Med. 2017, 6, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon-Shiong, P.; Heintz, R.E.; Merideth, N.; Yao, Q.X.; Yao, Z.; Zheng, T.; Murphy, M.; Moloney, M.K.; Schmehl, M.; Harris, M.; et al. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet 1994, 343, 950–951. [Google Scholar] [CrossRef]
- Soon-Shiong, P.; Feldman, E.; Nelson, R.; Heintz, R.; Yao, Q.; Yao, Z.; Zheng, T.; Merideth, N.; Skjak-Braek, G.; Espevik, T. Long-term reversal of diabetes by the injection of immunoprotected islets. Proc. Natl. Acad. Sci. USA 1993, 90, 5843–5847. [Google Scholar] [CrossRef] [PubMed]
- Strand, B.L.; Ryan, T.L.; In’t Veld, P.; Kulseng, B.; Rokstad, A.M.; Skjak-Brek, G.; Espevik, T. Poly-L-Lysine induces fibrosis on alginate microcapsules via the induction of cytokines. Cell Transplant. 2001, 10, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.S.; Cruise, G.M.; Hager, S.R.; Lamberti, F.V.; Yu, X.; Garufis, C.L.; Yu, Y.; Mundwiler, K.E.; Cole, J.F.; Hubbell, J.A.; et al. Immunoisolation of adult porcine islets for the treatment of diabetes mellitus. The use of photopolymerizable polyethylene glycol in the conformal coating of mass-isolated porcine islets. Ann. N. Y. Acad. Sci. 1997, 831, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Kızılel, S. Theoretical and Experimental Investigation for Interfacial Photopolymerization of Poly(ethylene glycol) Diacrylate. Ph.D. Thesis, Illinois Institute of Technology, Chicago, IL, USA, 2004. [Google Scholar]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Mathematical model for surface-initiated photopolymerization of poly(ethylene glycol) diacrylate. Macromol. Theory Simul. 2006, 15, 686–700. [Google Scholar] [CrossRef]
- Betre, H.; Setton, L.A.; Meyer, D.E.; Chilkoti, A. Characterization of a genetically engineered elastin-like polypeptide for cartilaginous tissue repair. Biomacromolecules 2002, 3, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S.; Lee, D.A.; Bader, D.L. Crosslinking density influences the morphology of chondrocytes photoencapsulated in PEG hydrogels during the application of compressive strain. J. Orthop. Res. 2004, 22, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, S.J.; Anseth, K.S. Controlling the spatial distribution of ECM components in degradable PEG hydrogels for tissue engineering cartilage. J. Biomed. Mater. Res. Part A 2003, 64, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S. Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly(ethylene glycol) hydrogels. J. Biomed. Mater. Res. 2002, 59, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Nuttelman, C.R.; Anseth, K.S. The effects of crosslinking density on cartilage formation in photocrosslinkable hydrogels. Biomed. Sci. Instrum. 1999, 35, 309–314. [Google Scholar] [PubMed]
- Bidarra, S.J.; Barrias, C.C.; Granja, P.L. Injectable alginate hydrogels for cell delivery in tissue engineering. Acta Biomater. 2014, 10, 1646–1662. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Diniz, I.M.; Chen, C.; Aghaloo, T.; Wu, B.M.; Shi, S.; Moshaverinia, A. Alginate/hyaluronic acid hydrogel delivery system characteristics regulate the differentiation of periodontal ligament stem cells toward chondrogenic lineage. J. Mater. Sci. Mater. Med. 2017, 28, 162. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S. The effects of scaffold thickness on tissue engineered cartilage in photocrosslinked poly(ethylene oxide) hydrogels. Biomaterials 2001, 22, 619–626. [Google Scholar] [CrossRef]
- Nuttelman, C.R.; Benoit, D.S.W.; Tripodi, M.C.; Anseth, K.S. The effect of ethylene glycol methacrylate phosphate in PEG hydrogels on mineralization and viability of encapsulated hMSCs. Biomaterials 2006, 27, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. J. Biomed. Mater. Res. Part A 2006, 76, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. In vitro osteogenic differentiation of human mesenchymal stem cells photoencapsulated in PEG hydrogels. J. Biomed. Mater. Res. Part A 2004, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Betre, H.; Ong, S.R.; Guilak, F.; Chilkoti, A.; Fermor, B.; Setton, L.A. Chondrocytic differentiation of human adipose-derived adult stem cells in elastin-like polypeptide. Biomaterials 2005, 27, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, L.; Sun, M.; Zhang, Y.; Chen, L.; Rong, Y.; Li, Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 2015, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Arhaa, M.; Choudhary, S.; Ashton, R.S.; Bhatia, S.R.; Schaffer, D.V.; Kane, R.S. The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 2009, 30, 4695–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldshmid, R.; Seliktar, D. Hydrogel Modulus Affects Proliferation Rate and Pluripotency of Human Mesenchymal Stem Cells Grown in Three-Dimensional Culture. ACS Biomater. Sci. Eng. 2017, 7, 3433–3446. [Google Scholar] [CrossRef]
- Alakpa, E.V.; Jayawarna, V.; Lampel, A.; Burgess, K.V.; West, C.C.; Bakker, S.C.J.; Roy, S.; Javid, N.; Fleming, S.; Lamprou, D.A.; et al. Tunable Supramolecular Hydrogels for Selection of Lineage-Guiding Metabolites in Stem Cell Cultures. Chem 2016, 1, 298–319. [Google Scholar] [CrossRef] [Green Version]
- Lanniel, M.; Huq, E.; Allen, S.; Buttery, L.; Williams, P.M.; Alexander, M.R. Substrate induced differentiation of human mesenchymal stem cells on hydrogels with modified surface chemistry and controlled modulus. Soft Matter 2011, 7, 6501–6514. [Google Scholar] [CrossRef]
- Hasan, A.; Khattab, A.; Islam, M.A.; Hweij, K.A.; Zeitouny, J.; Waters, R.; Sayegh, M.; Hossain, M.M.; Paul, A. Injectable Hydrogels for Cardiac Tissue Repair after Myocardial Infarction. Adv. Sci. 2015, 2, 1500122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, M.; Vaibavi, S.R.; Rufaihah, A.J.; Nithya, V.; Wang, J.; Shachaf, Y.; Kofidis, T.; Seliktar, D. The effect of matrix stiffness of injectable hydrogels on the preservation of cardiac function after a heart attack. Biomaterials 2014, 35, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Kim, I.K.; Cho, S.W.; Cho, M.C.; Hwang, K.K.; Piao, H.; Piao, S.; Lim, S.H.; Hong, Y.S.; Choi, C.Y.; et al. Implantation of bone marrow mononuclear cells using injectable fibrin matrix enhances neovascularization in infarcted myocardium. Biomaterials 2005, 26, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hu, Q.; Braunlin, E.A.; Suggs, L.J.; Zhang, J. Enhancing efficacy of stem cell transplantation to the heart with a PEGylated fibrin biomatrix. Tissue Eng. Part A 2008, 14, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zheng, J.; Wang, H.; Becker, M.L.; Leipzig, N.D. Neural stem cell encapsulation and differentiation in strain promoted crosslinked polyethylene glycol-based hydrogels. J. Biomater. Appl. 2018, 32, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wijekoon, A.; Leipzig, N.D. 3D Differentiation of Neural Stem Cells in Macroporous Photopolymerizable Hydrogel Scaffolds. PLoS ONE 2012, 7, e48824. [Google Scholar] [CrossRef] [PubMed]
- Aurand, E.R.; Wagner, J.L.; Shandas, R.; Bjugstad, K.B. Hydrogel formulation determines cell fate of fetal and adult neural progenitor cells. Stem Cell Res. 2014, 12, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Zisch, A.H.; Lutolf, M.P.; Ehrbar, M.; Raeber, G.P.; Rizzi, S.C.; Davies, N.; Schmoekel, H.; Bezuidenhout, D.; Djonov, V.; Zilla, P.; et al. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 2003, 17, 2260–2262. [Google Scholar] [CrossRef] [PubMed]
- Masters, K.S.; Shah, D.N.; Walker, G.; Leinwand, L.A.; Anseth, K.S. Designing scaffolds for valvular interstitial cells: Cell adhesion and function on naturally derived materials. J. Biomed. Mater. Res. Part A 2004, 71, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-G.; Itle, L.J.; Pishko, M.V. Molding of hydrogel microstructures to create multiphenotype cell microarrays. Anal. Chem. 2003, 75, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Zguris, J.C.; Itle, L.J.; Koh, W.-G.; Pishko, M.V. A Novel Single-Step Fabrication Technique to Create Heterogeneous Poly(ethylene glycol) Hydrogel Microstructures Containing Multiple Phenotypes of Mammalian Cells. Langmuir 2005, 21, 4168–4174. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-G.; Pishko, M. Photoreaction Injection Molding of Biomaterial Microstructures. Langmuir 2003, 19, 10310–10316. [Google Scholar] [CrossRef]
- Zisch, A.H.; Lutolf, M.P.; Hubbell, J.A. Biopolymeric delivery matrices for angiogenic growth factors. Cardiovasc. Pathol. 2003, 12, 295–310. [Google Scholar] [CrossRef]
- Burdick, J.A.; Mason, M.N.; Hinman, A.D.; Thorne, K.; Anseth, K.S. Delivery of osteoinductive growth factors from degradable PEG hydrogels influences osteoblast differentiation and mineralization. J. Control. Release 2002, 83, 53–63. [Google Scholar] [CrossRef]
- Zisch, A.H.; Zeisberger, S.M.; Ehrbar, M.; Djonov, V.; Weber, C.C.; Ziemiecki, A.; Pasquale, E.B.; Hubbell, J.A. Engineered fibrin matrixes for functional display of cell membrane-bound growth factor-like activities: Study of angiogenic signaling by ephrin-B2. Biomaterials 2004, 25, 3245–3257. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.K.A.; Richard, C.; Bessodes, M.; Scherman, D.; Merten, O.W. Growth Factor Delivery Approaches in Hydrogels. Biomacromolecules 2009, 10, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Ward, M.; Liang, E.; Young, M.J.; Langer, R. Stimulation of neurite outgrowth by neurotrophins delivered from degradable hydrogels. Biomaterials 2006, 27, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kang, Y.M.; Kang, K.N.; Kim, D.Y.; Kim, H.J.; Min, B.H.; Kim, J.H.; Kim, M.S. Wound Dressings for Wound Healing and Drug Delivery. Tissue Eng. Regener. Med. 2011, 8, 1–7. [Google Scholar]
- Itle, L.J.; Koh, W.-G.; Pishko, M.V. Multi-phenotypic cellular arrays for biosensing. BioMEMS Biomed. Nanotechnol. 2006, 3, 79–93. [Google Scholar]
- Pishko, M.V. Cell based bioassays in microfluidic systems. Polym. Prepr. 2006, 47, 1076–1077. [Google Scholar]
- Hoffman, A.S.; Stayton, P.S.; Bulmus, V.; Chen, G.; Chen, J.; Cheung, C.; Chilkoti, A.; Ding, Z.; Dong, L.; Fong, R.; et al. Founder’s Award, Society for Biomaterials. Sixth World Biomaterials Congress 2000, Kamuela, HI, May 15–20, 2000. Really smart bioconjugates of smart polymers and receptor proteins. J. Biomed. Mater. Res. 2000, 52, 577–586. [Google Scholar] [CrossRef]
- Itle, L.J.; Zguris, J.C.; Pishko, M.V. Cell-based bioassays in microfluidic systems. Proc. SPIE Int. Soc. Opt. Eng. 2004, 5588, 9–18. [Google Scholar]
- Kulkarni, R.V.; Biswanath, S. Electrically responsive smart hydrogels in drug delivery: A review. J. Appl. Biomater. Biomech. 2007, 5, 125–139. [Google Scholar] [PubMed]
- Velasco, D.; Elvira, C.; San Roman, J. New stimuli-responsive polymers derived from morpholine and pyrrolidine. J. Mater. Sci. Mater. Med. 2008, 19, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Prabaharan, M.; Mano, J.F. Stimuli-responsive hydrogels based on polysaccharides incorporated with thermo-responsive polymers as novel biomaterials. Macromol. Biosci. 2006, 6, 991–1008. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Luna, V.H.; González-Reynoso, O. Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications. Gels 2018, 4, 61. https://doi.org/10.3390/gels4030061
Pérez-Luna VH, González-Reynoso O. Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications. Gels. 2018; 4(3):61. https://doi.org/10.3390/gels4030061
Chicago/Turabian StylePérez-Luna, Víctor H., and Orfil González-Reynoso. 2018. "Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications" Gels 4, no. 3: 61. https://doi.org/10.3390/gels4030061
APA StylePérez-Luna, V. H., & González-Reynoso, O. (2018). Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications. Gels, 4(3), 61. https://doi.org/10.3390/gels4030061