On Going to a New Era of Microgel Exhibiting Volume Phase Transition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
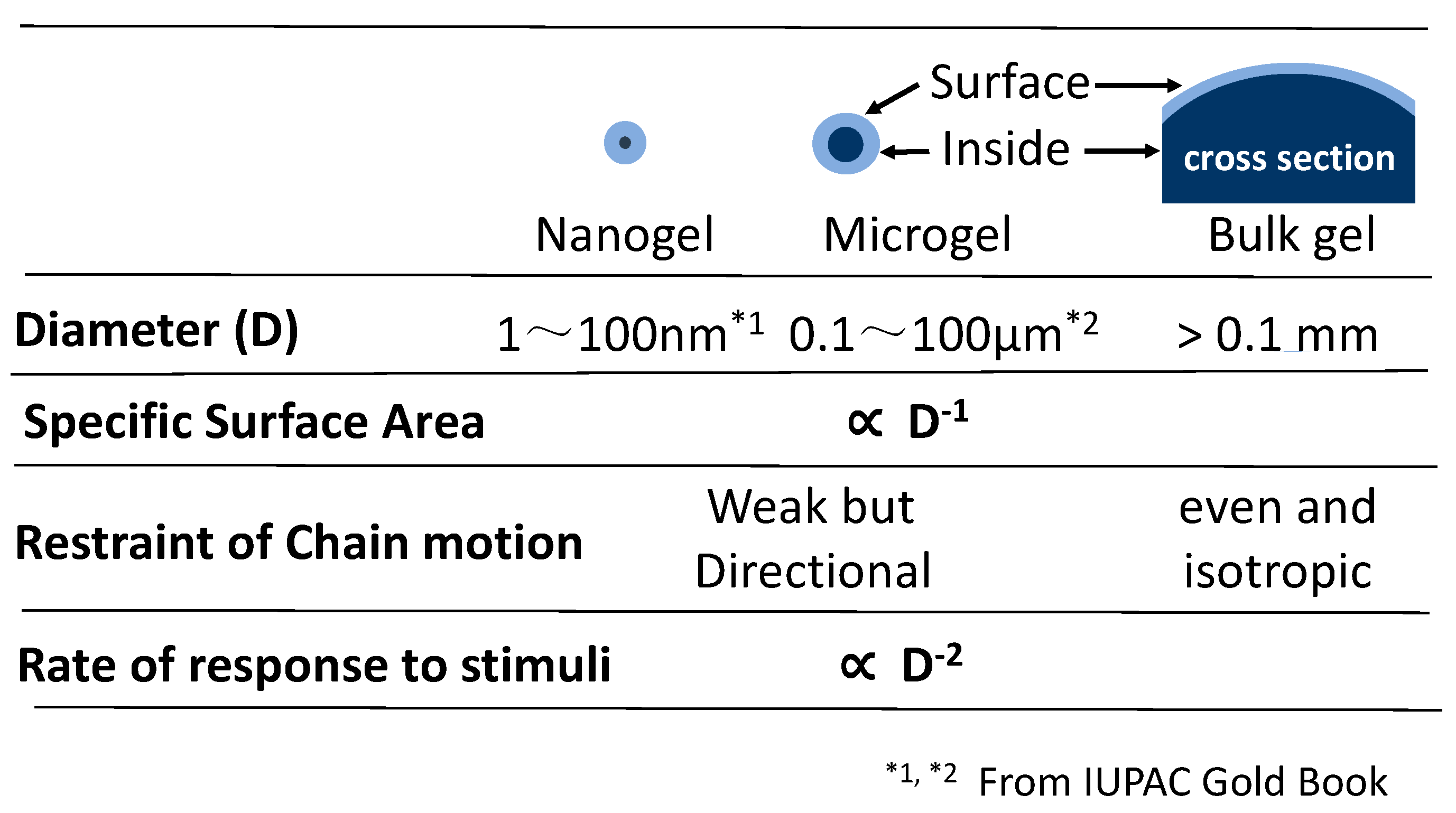
1.1. Definition of Nanogel and Microgel
1.2. Comparison of Microgels and Bulk Gels
1.3. Volume Phase Transition of Microgels
2. Synthesis of Microgels Exhibiting VPT
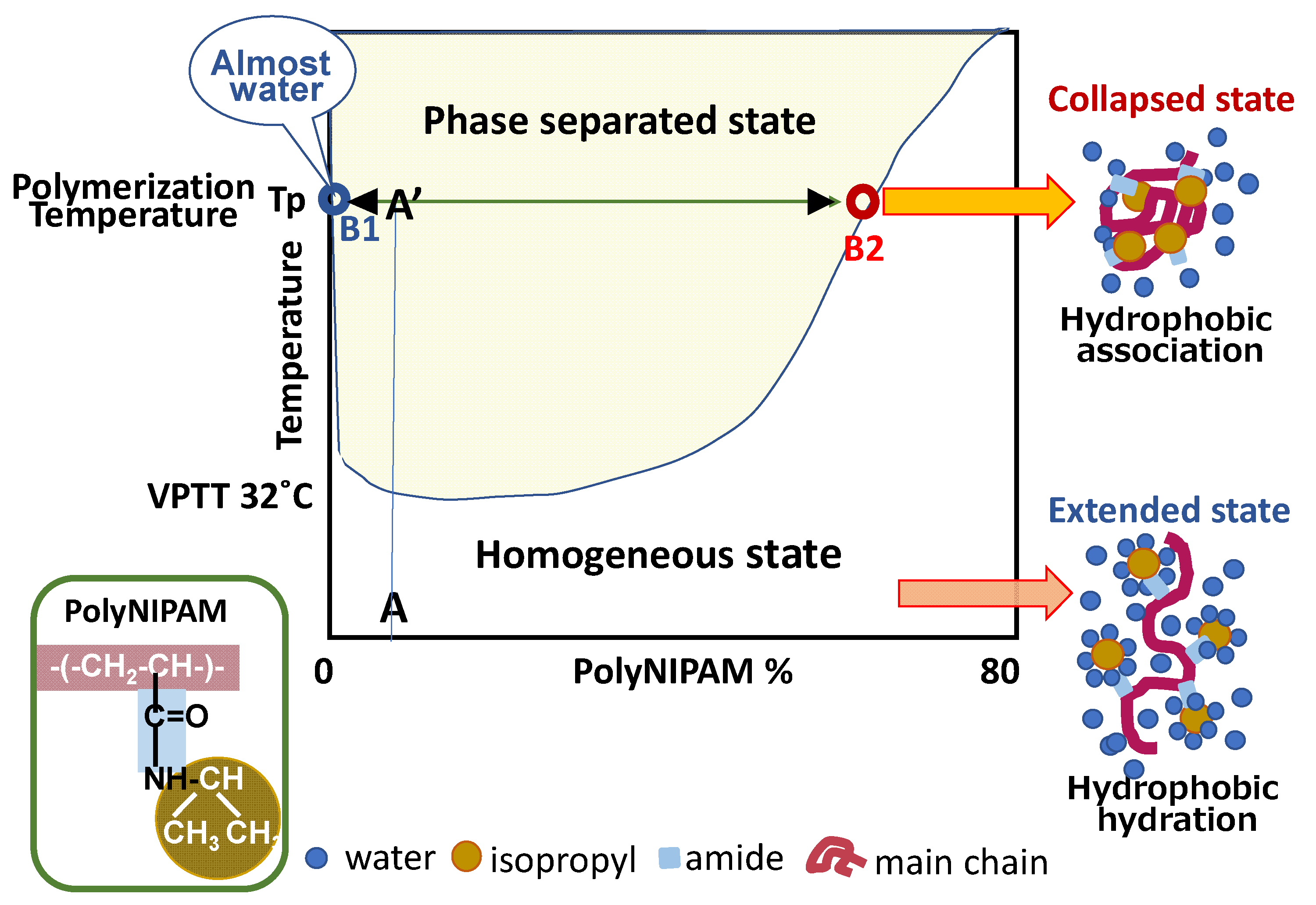
2.1. Precipitation Polymerization Elucidated from Phase Diagram
2.2. Mechanism and Kinetics of Precipitation Polymerization
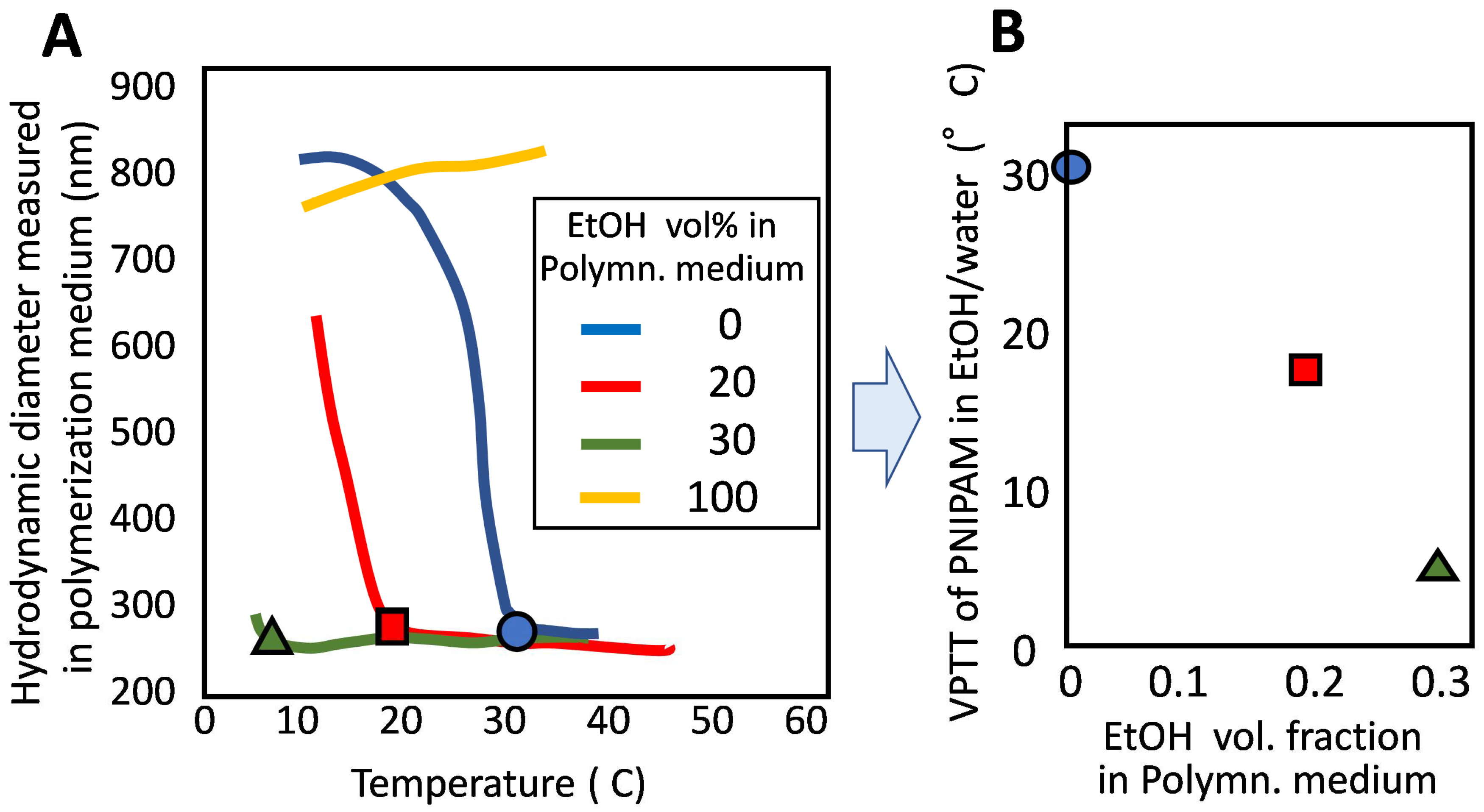
2.3. Synthesis of Microgels and Their VPT Behavior
2.3.1. Microgels Prepared by Precipitation Polymerization and Their VPT
2.3.2. Other Techniques to Prepare Thermosensitive Microgels
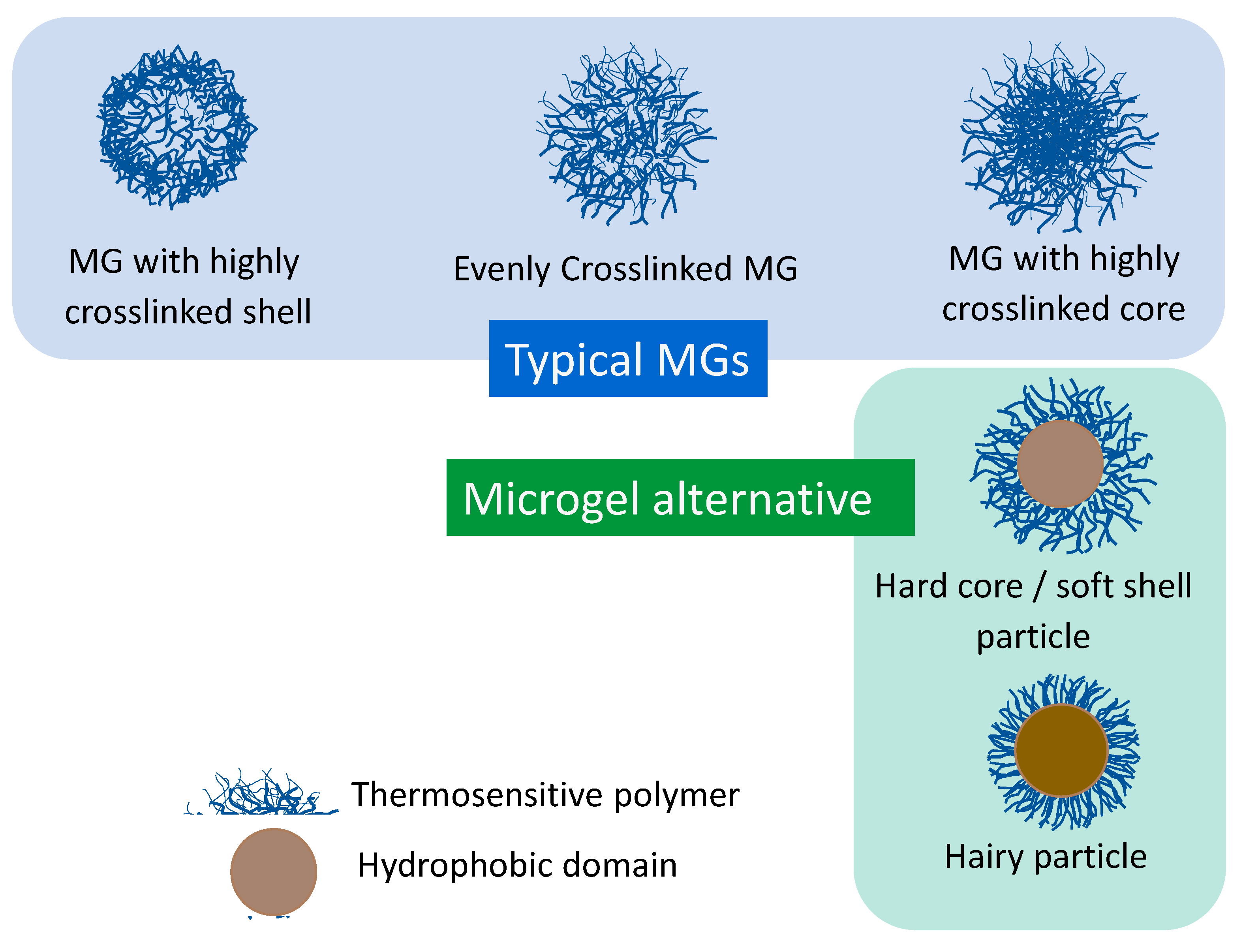
2.4. Synthesis and Characteristics of Microgel Alternatives
2.4.1. Hard Core/Soft Shell Microspheres Exhibiting VPT
2.4.2. Hairy Microspheres Exhibiting VPT
3. Design of Thermosensitive Microgels
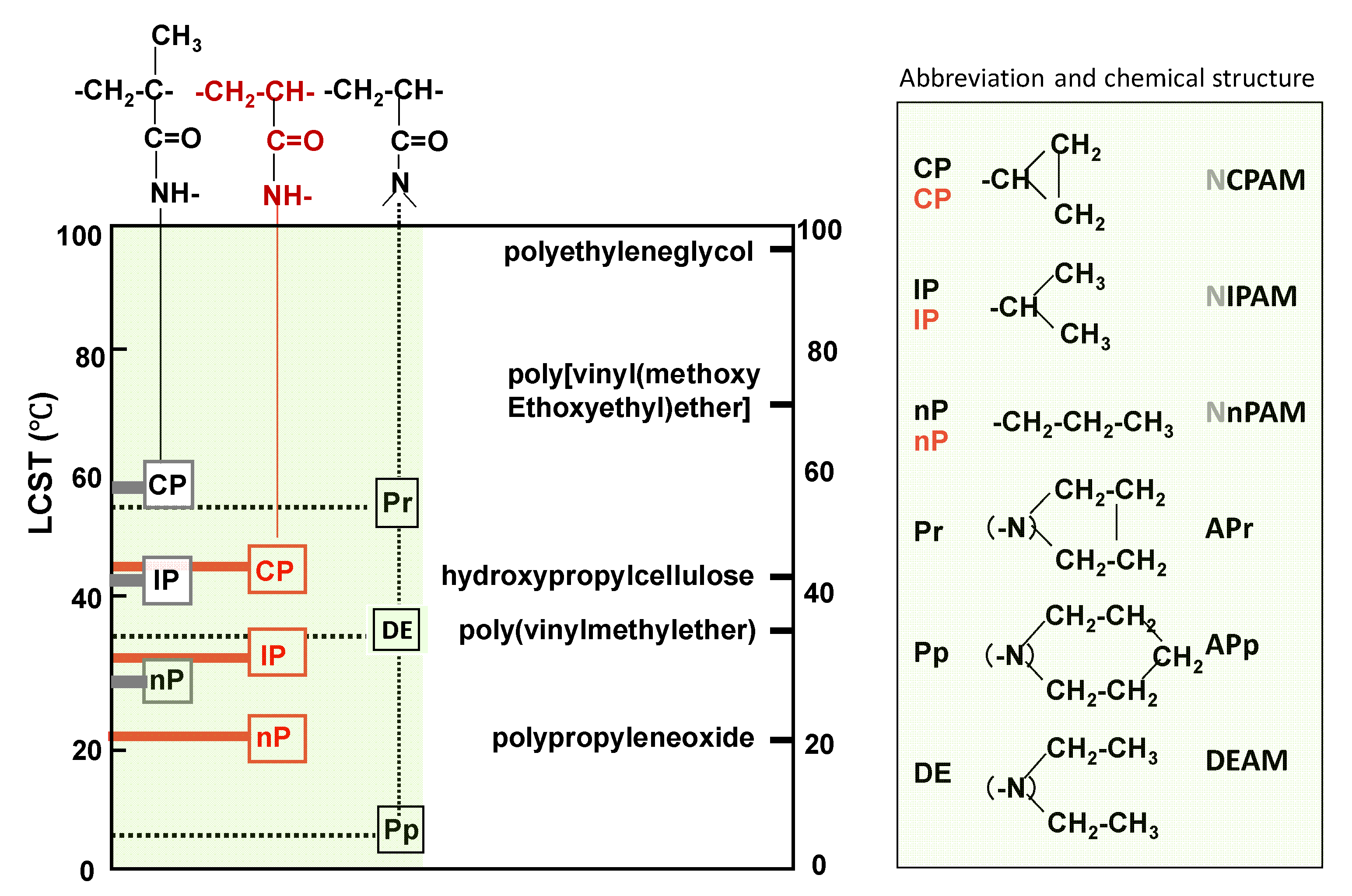
3.1. VPTT and Its Sharpness of Microgels
3.2. Size and Its Distribution of Microgels
3.3. Cross-Linking Density and Its Distribution
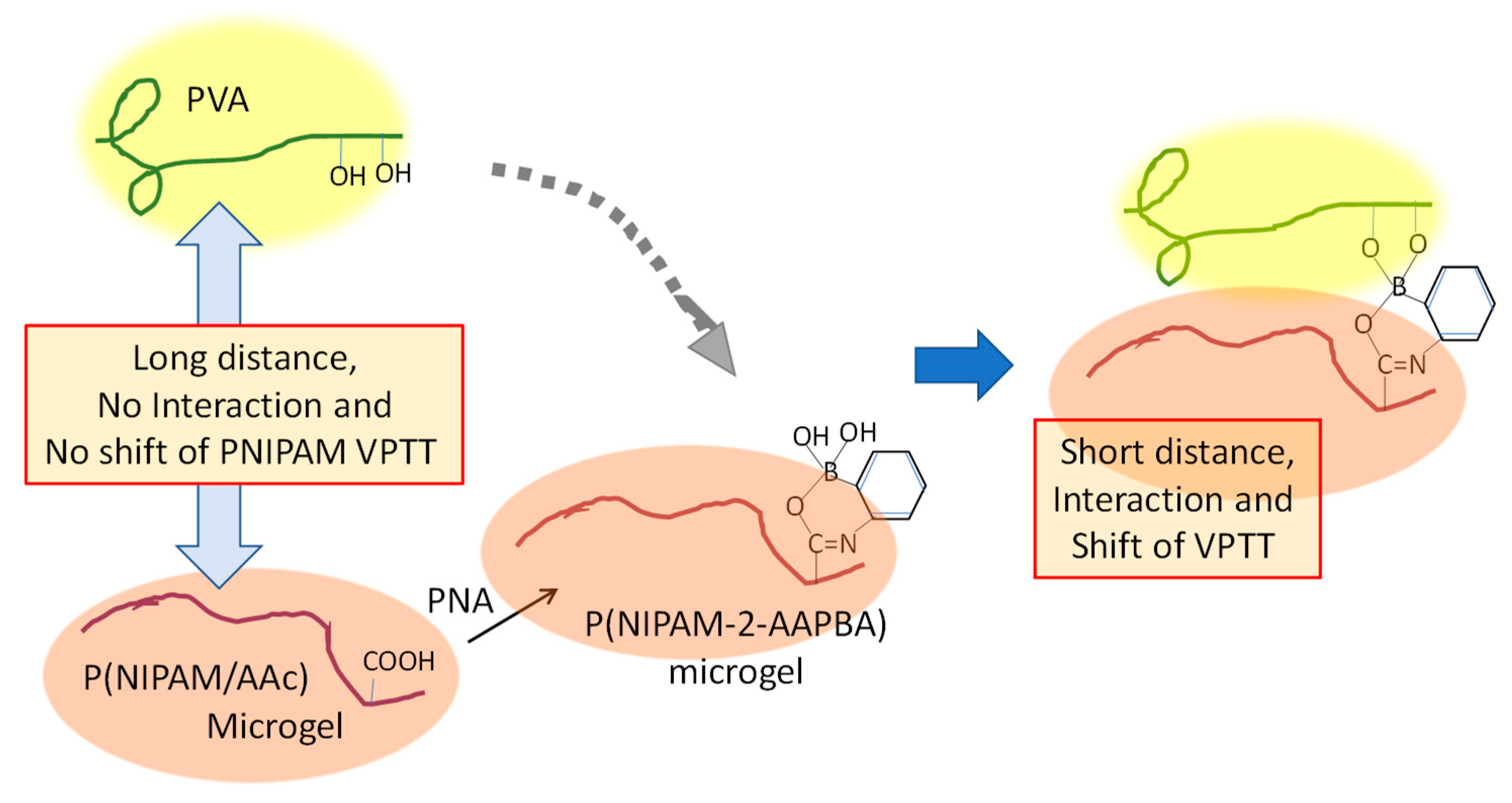
3.4. Hybridization
4. Physicochemical Characteristics of Thermosensitive Microgels
4.1. Surface Activity and Adsorption of Microgels
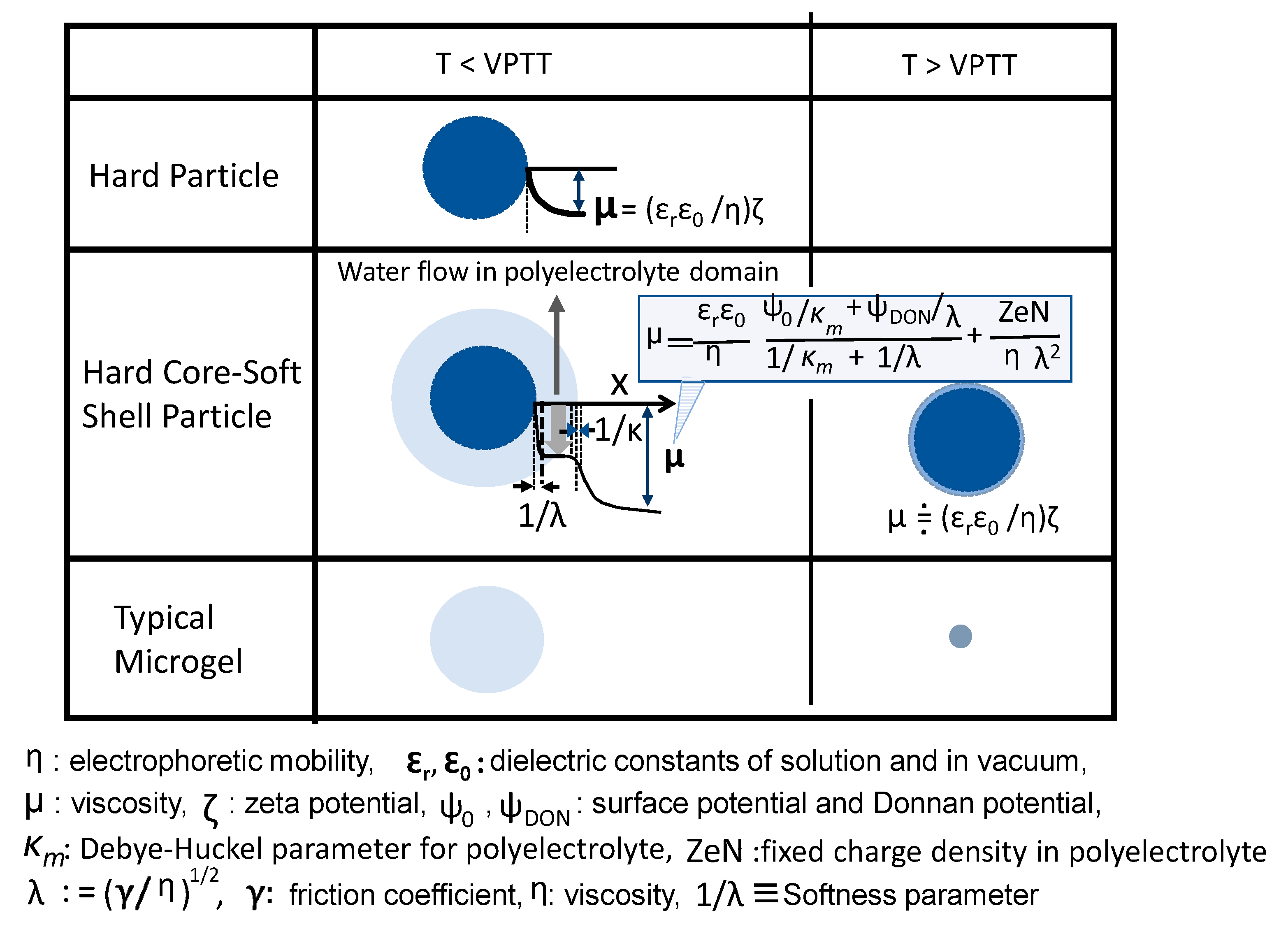
4.2. Electrical Surface Phenomenon of Microgels
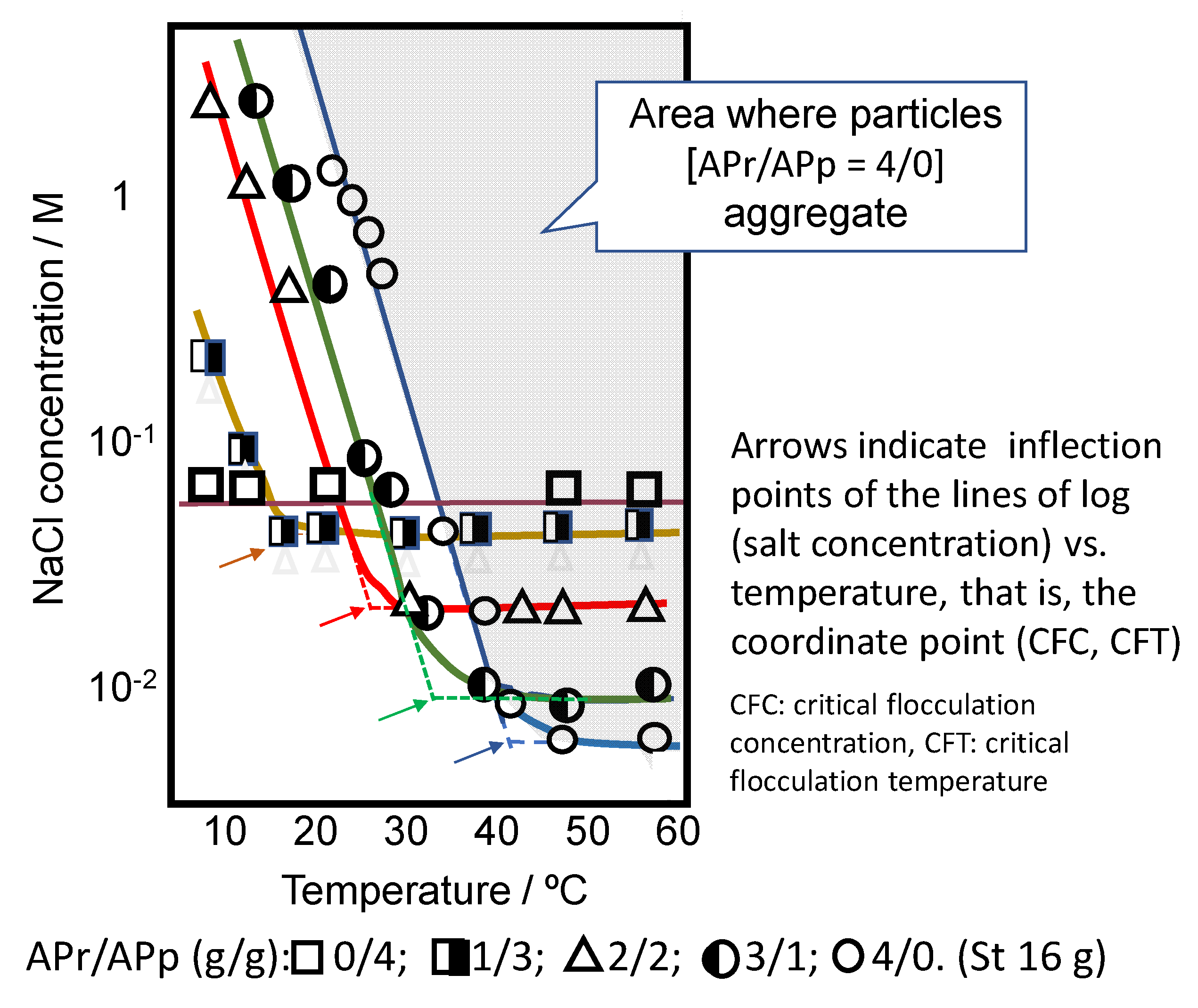
4.3. Dispersion and Aggregation of Microgels
5. Remaining Problems on Functional Microgels and Strategies for Solving Them
6. Afterword
Funding
Conflicts of Interest
References
- Tanaka, T. Collapse of gels and the critical endpoint. Phys. Rev. Lett. 1978, 40, 820–823. [Google Scholar] [CrossRef]
- Pelton, R. Temperature-Sensitive aqueous microgels. Adv. Colloid Interface Sci. 2000, 85, 1–32. [Google Scholar] [CrossRef]
- Nayak, S.; Lyon, L.A. Soft nanotechnology with soft nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 7686–7708. [Google Scholar] [CrossRef] [PubMed]
- Karg, M.; Pich, A.; Hellweg, T.; Hoare, T.; Lyon, L.A.; Crssous, J.J.; Suzuki, D.; Gumerov, R.A.; Scheider, S.; Potemkin, I.I.; et al. Nanogels and microgels: From model colloids to applications, recent developments, and future trends. Langmuir 2019, 35, 6231–6255. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Koga, T.; Winnik, F.M. Temperature-Responsive polymers in mixed solvents: Competitive hydrogen bonds cause cononsolvency. Phys. Rev. Lett. 2008, 101, 028302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, F.; Okada, Y. Theoretical study on the phase diagrams of associating Polymers, I. Non-Gelling system. Netsu Sokutei 2005, 32, 178. [Google Scholar]
- Maeda, Y.; Higuchi, T.; Ikeda, I. Change in hydration state during the coil-globule transition of aqueous solutions of (N-isopropylacrylamide) as evidenced by FTIR. Langmuir 2000, 16, 7503–7509. [Google Scholar] [CrossRef]
- Maeda, Y.; Nakajima, T.; Ikeda, I. Changes in the hydration states of poly(N-alkylacrylamide)s during their phase transitions in water observed by FTIR spectroscopy. Macromolecules 2001, 34, 1381–1399. [Google Scholar] [CrossRef]
- Matsumura, Y.; Iwai, K. Thermo-Responsive behavior and microenvironments of poly(N-isopropylacrylamide) microgel particles as studied by fluorescent label method. J. Colloid Interface Sci. 2006, 296, 102–109. [Google Scholar] [CrossRef]
- Sbeih, S.; Mohanty, P.S.; Morrow, M.R.; Yethiraj, A. Structural parameters of soft PNIPAM microgel particles as a function of crosslink density. J. Colloid Interface Sci. 2019, 552, 781–793. [Google Scholar] [CrossRef]
- De Oliveira, T.E.; Marques, C.M.; Netz, P.A. Molecular dynamics study of the LCST transition in aqueous poly(N-n-propylacrylamide). Phys. Chem. Chem. Phys. 2018, 20, 10100–10107. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Ino, H.; Maie KNakayama, M.; Kano, A.; Maruyama, A. Ureido-Derivatized Polymers based on both Poly(allylurea) and poly(l-citrulline) exhibit UCST-type phase transition behavior under physiologically relevant conditions. Biomacromolecules 2011, 12, 3418–3422. [Google Scholar] [CrossRef] [PubMed]
- Heskins, M.; Guillet, J.E. Solution properties of poly(N-isopropylacrylamide). J. Macromol. Sci. A 1968, 2, 1441–1455. [Google Scholar] [CrossRef]
- Pelton, R. Poly(N-isopropylacrylamide) (PNIPAM) is never hydrophobic. J. Colloid Interface Sci. 2010, 348, 673–674. [Google Scholar] [CrossRef]
- Tauer, K.; Gau, D.; Schulze, S.; Voklel, A.; Dimova, R. Thermal property changes of poly(N-isopropylacrylamide) microgel particles and block copolymers. Colloid Polym. Sci. 2009, 287, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Pelton, R.H.; Hamielec, A.E.; Woods, D.R.; McPhee, W. The kinetics of poly(N-isopropylacrylamide) microgel latex formation. Colloid Polym. Sci. 1994, 272, 467–477. [Google Scholar] [CrossRef]
- Virtanen, O.L.J.; Richitering, W. Kinetics and particle size control in non-stirred precipitation polymerization of N-isopropylacrylamide. Colloid Polym. Sci. 2014, 292, 1743–1756. [Google Scholar] [CrossRef]
- Fukai, T.; Shinyashiki, N.; Yagihara, S.; Kit, R.; Tanaka, F. Phase behavior of co-nonsolvent systems: Poly(N-isopropylacrylamide) in mixed solvents of water and methanol. Langmuir 2018, 34, 3003–3009. [Google Scholar] [CrossRef]
- Costa, R.O.; Freitas, R.F. Phase behavior of poly(N-isopropylacrylamide) in binary aqueous solutions. Polymer 2002, 43, 5879–5885. [Google Scholar] [CrossRef]
- Takahashi, T.; Fukazawa, H.; Kawaguchi, K. Particle-forming precipitation polymerization under unusual conditions. Prog. Colloid Polym. Sci. 2003, 124, 164–167. [Google Scholar]
- Pelton, R.H.; Chibante, P. Preparation of aqueous latices with N-isopropylacrylamide. Colloids Surf. 1986, 20, 247–256. [Google Scholar] [CrossRef]
- Pelton, R.H.; Pelton, H.M.; Morphesis, A.; Rowell, R.L. Particle sizes and electrophoretic mobilities of poly(N-isopropylacrylamide) latex. Langmuir 1989, 5, 816–818. [Google Scholar] [CrossRef]
- Jones, C.D.; Lyon, L.A. Shell-restricted swelling and core compression in poly(N-isopropylacrylamide) core-shell microgels. Macromolecules 2003, 36, 1988–1993. [Google Scholar] [CrossRef]
- Berndt, I.; Pedersen, J.S.; Lindner, P.; Richtering, W. Structure of doubly temperature sensitive core-shell microgels based on poly-N-isopropylacrylamide and poly-Nisopropyl- methacrylamide. Prog. Colloid Polym. Sci. 2006, 133, 35–40. [Google Scholar]
- Belndt, I.; Pederson, J.S.; Richtering, W. Structure of multi-responsive “intelligent” core-shell microgels. JACS 2005, 127, 9372–9373. [Google Scholar] [CrossRef]
- Berndt, I.; Popescu, C.; Wortmann, F.J.; Richtering, W. Mechanics versus thermodynamics: Swelling in multiple temperature sensitive core-shell microgels. Angew. Chem. Int. Ed. 2006, 45, 1081–1085. [Google Scholar] [CrossRef]
- Keerl, M.; Richtering, W. Synergistic depression of volume phase transition temperature in copolymer microgels. Colloid Polym. Sci. 2007, 285, 471–474. [Google Scholar] [CrossRef]
- Marian, M.; Wrede, O.; Wiehemeier, L.; Feoktstov, A.; Cousin, F.; Hellweg, T.; Oberdisse, J. Spatial distribution of core monomers in acrylamide-based core-shell microgels with linear swelling behaviour. Sci. Rep. 2019, 9, 13812. [Google Scholar]
- Jones, C.D.L.; Lyon, L.A. Synthesis and characterization of multiresponsive core-shell microgels. Macromolecules 2000, 33, 8301–8306. [Google Scholar] [CrossRef]
- Keeri, M.; Smirnovas, V.; Winter, R.; Richtering, W. Interplay between hydrogen bonding and macromolecular architecture leading to unusual phase behavior in thermosensitive microgels. Angew. Chem. Int. Ed. 2008, 47, 338–341. [Google Scholar]
- Fernandez, V.V.A.; Tepale, N.; Sánchez-Díaz, J.C. Thermoresponsive nanostructured poly(N-isopropylacrylamide) hydrogels made via inverse microemulsion polymerization. Colloid Polym. Sci. 2006, 284, 387–395. [Google Scholar] [CrossRef]
- Dowding, P.J.; Vincent, B.; Williams, E. Preparation and swelling properties of Poly(NIPAM) “Minigel” particles prepared by inverse suspension polymerization. J. Colloid Interface Sci. 2000, 221, 268. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.K.; Kim, J.W.; Agrest, J.J.; Weitz, D.A.; Chu, L.Y. Fabrication of monodisperse thermosensitive microgels and gel capsules in microfluidic devices. Soft Matter 2008, 4, 2303–2309. [Google Scholar] [CrossRef]
- Hua, M.; Du, Y.J.; Song, J.; Sun, M. Surfactant-Free fabrication of pNIPAAm microgels in microfluidic devices. J. Mater. Res. 2019, 14, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.W.; Wang, J.; Gao, W.; Jiao, J.; Feng, H.J.; Liu, X.; Chen, L.P. One-Pot preparation of thermo-responsive silica-poly(N-isopropylacrylamide) nanocomposite particles in supercritical carbon dioxide. J. Colloid Interface Sci. 2010, 343, 141–148. [Google Scholar] [CrossRef]
- Kojima, H. Studies on the phase transition of hydrogels and aqueous solutions of thermosensitive polymers. Polym. J. 2018, 50, 411–418. [Google Scholar] [CrossRef]
- Hoshino, F.; Kawaguchi, H.; Ohtsuka, Y. N-Substituted acrylamide-styrene copolymer latices III. Morphology of latex particles. Polym. J. 1987, 19, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, F.; Fujimoto, T.; Kawaguchi, H.; Ohtsuka, Y. N-Substituted acrylamide-styrene copolymer latices II. Polymerization behavior and thermosensitive stability of latices. Polym. J. 1987, 19, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Liu, L.; Yang, W. Preparation of soft hydrogel nanoparticles with PNIPAm hair and characterization of their temperature-induced aggregation. Langmuir 2010, 26, 2076–2082. [Google Scholar] [CrossRef]
- Li, X.; Yang, B.; Jia, X.; Chen, M.; Hu, Z. Temperature-Responsive hairy particle-supported proline for direct asymmetric aldol reaction in water. RSC Adv. 2015, 5, 89149–89156. [Google Scholar] [CrossRef]
- Tsuji, S.; Kawaguchi, H. Temperature-sensitive hairy particles prepared by living radical graft polymerization. Langmuir 2004, 20, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Kawaguchi, H. Effect of graft chain length and structure design on temperature-sensitive hairy particles. Macromolecules 2006, 39, 4338–4344. [Google Scholar] [CrossRef]
- Ito, S. Thermosensitive polyacrylamide derivatives. Netsu Sokutei 1992, 19, 9193. [Google Scholar]
- Fujishige, S.; Kubota, K.; Ando, I. Phase transition of aqueous solutions of poly(N-isopropylacrylamide) and poly(N-isopropylmethacrylamide). J. Phys. Chem. 1989, 93, 3311–3313. [Google Scholar] [CrossRef]
- Kano, M.; Kokufuta, E. On the temperature-responsive polymers and gels based on N-Propylacrylamides and N-Propylmethacrylamides. Langmuir 2009, 25, 8649–8655. [Google Scholar] [CrossRef]
- Tang, Z.J.; Weng, Y.; Guan, Y.; Zhang, Y.G. Unexpected large depression of VPTT of a PNIPAM microgel by low concentration of PVA. Macromol. Chem. Phys. 2017, 218, 201700364. [Google Scholar] [CrossRef]
- Zhu, P.W.; Napper, D.H. Experimental observation of coil-to-globule type transitions at interfaces. J. Colloid Interface Sci. 1994, 164, 489. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, S. Volume phase transition of swollen gels: Discontinuous or continuous? Macromolecules 1997, 30, 574–576. [Google Scholar] [CrossRef]
- Matsuoka, H.; Fujimoto, K.; Kawaguchi, H. Monodisperse microspheres exhibiting discontinuous response to temperature change. Polym. Gels Netw. 1999, 6, 319–332. [Google Scholar] [CrossRef]
- Kardos, A.; Gilányi, T.; Varga, I. How small can poly(N-isopropylacrylamide) nanogels be prepared by controlling the size with surfactant? J. Colloid Interface Sci. 2019, 557, 793–806. [Google Scholar] [CrossRef]
- Shimizu, H.; Wada, R.; Okabe, M. Preparation and Characterization of Micrometer-Sized Poly(N-isopropylacrylamide) Hydrogel Particles. Polym. J. 2009, 41, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Smith, M.H.; Lyon, L.A. Temperature-Programmed synthesis of micron-sized multi-responsive microgels. Colloid Polym. Sci. 2009, 287, 277–285. [Google Scholar] [CrossRef]
- Hu, X.; Tong, Z.; Lyon, L.A. Control of Poly(N-isopropylacrylamide) microgel network structure by precipitation polymerization near the lower critical solution temperature. Langmuir 2011, 27, 4142–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; MKwok, H.; Ngai, T. Preparation of responsive micrometer-sized microgel particles with a highly functionalized shell. Rapid Commun. 2012, 33, 419–425. [Google Scholar] [CrossRef]
- Kwok, M.H.; Li, Z.F.; Ngai, T. Controlling the synthesis and characterization of micrometer-sized PNIPAM microgels with tailored morphologies. Langmuir 2013, 29, 9581–9591. [Google Scholar] [CrossRef]
- Minato, H.; Murai, M.; Watanabe, T.; Matsui, S.; Takizawa, M.; Kureha, T.; Suzuki, D. The deformation of hydrogel microspheres at the air/water interface. Chem. Commun. 2018, 54, 32–35. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Matsui, S.; Kureha, T.; Shibayama, M.; Uchihashi, T.; Suzuki, D. Non-Thermoresponsive decanano-sized domains in thermoresponsive hydrogel microspheres revealed by temperature-controlled high-speed atomic force microscopy. Angew. Chem. Int. Ed. 2019, 58, 8809–8813. [Google Scholar] [CrossRef]
- Virtanen, O.L.J.; Mourran, A.; Pinard, P.I.; Richitering, W. Persulfate initiated ultra-low cross-linked poly(N-isopropylacrylamide) microgels possess an unusual inverted cross-linking structure. Soft Matter 2016, 12, 3919–3928. [Google Scholar] [CrossRef]
- Gao, J.; Frisken, B.J. Cross-Linker free N-isopropylacrylamide gel nanospheres. Langmuir 2003, 19, 5212–5216. [Google Scholar] [CrossRef]
- Zhang, J.; Pelton, R. Poly(N-isopropylacrylamide) microgels at the air-water interface. Langmuir 1999, 15, 8032–8036. [Google Scholar] [CrossRef]
- Perez, P.; Gallardo, A.; Corringan, O.I.; Roman, J.S. Thermosensitivity of N-isopropylacrylamide hydrogels cross-linked with degradable cross-linker. J. Biomater. Sci. Polym. Ed. 2008, 19, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.M.; Liras, M.; Garrido, I.Q.; Gallardo, A.; Paris, R. Swelling control in thermo-responsive hydrogels based on 2-(2-methoxyethoxy)ethyl methacrylate by crosslinking and copolymerization with N-isopropylacrylamide. Polym. J. 2011, 43, 887–892. [Google Scholar] [CrossRef] [Green Version]
- Lorenzoab, F.D.; Seiffert, S. Nanostructural heterogeneity in polymer networks and gels. Polym. Chem. 2015, 6, 5515–5528. [Google Scholar] [CrossRef]
- Seiffert, S. Origin of nanostructural inhomogeneity in polymer-network gels. Polym. Chem. 2017, 8, 4443–4604. [Google Scholar] [CrossRef]
- Chai, S.; Zhang, J.; Yang, T.; Yuan, J.; Cheng, S. Thermoresponsive microgel decorated with silica nanoparticles in shell: Biomimetic synthesis and drug release application. Colloid Surf. A Physicochem. Eng. Asp. 2010, 356, 32–39. [Google Scholar] [CrossRef]
- Ballauff, M.; Lu, Y. “Smart” nanoparticles: Preparation, characterization and applications. Polymer 2007, 48, 1815–1823. [Google Scholar] [CrossRef] [Green Version]
- Butler, S.; Neogi, P.; Urban, B.; Fujita, Y.; Hu, Z.B.; Neogi, A. Zno nanoparticles in hydrogel polymer network for bio-imaging. Glob. J. Nanomed. 2017, 1, 555572. [Google Scholar]
- Cui, G.H.; Zhao, K.M.; You, K.W.; Gao, Z.G.; Kakuchi, T. Synthesis and characterization of phenylboronic acid-containing polymer for glucose-triggered drug delivery. Sci. Technol. Adv. Mater. 2020, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Hirose, Y.; Kato, T. Effects of temperature and concentration on surface tension of Poly(N-isopropylacrylamide) solutions. Langmuir 1996, 12, 3523–3526. [Google Scholar] [CrossRef]
- Monteux, C.; Marliere, C.; Paris, P.; Pantooustier, N.; Sanson, N.; Perrin, P. Poly(N-isopropylacrylamide) microgels at the oil-water interface: Interfacial properties as a function of temperature. Langmuir 2010, 26, 13839–13846. [Google Scholar] [CrossRef]
- Li, Z.F.; Richtering, W.; Ngai, T. Poly(N-isopropylacrylamide) microgels at the oil-water interface: Temperature effect. Soft Matter 2014, 10, 6182–6191. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.H.; Ngai, T. A confocal microscopy study of micron-sized poly(N-isopropylacrylamide) microgel particles at the oil–water interface and anisotopic flattening of highly swollen microgel. J. Colloid Interface Sci. 2016, 461, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Destibats, M.; Eyharts, M.; Lapeyre, V.; Sellier, E.; Varga, I.; Ravaine, V.; Scchmitt, V. Impact of pNIPAM microgel size on its ability to stabilize pickering emulsions. Langmuir 2014, 30, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Kawaguchi, H. Self-Assembly of Poly(N-isopropylacrylamide)-carrying microspheres into two-dimensional colloidal arrays. Langmuir 2005, 21, 2434–2437. [Google Scholar] [CrossRef]
- Tsuji, S.; Kawaguchi, H. Colored thin films prepared from hydrogel microspheres. Langmuir 2005, 21, 8439–8442. [Google Scholar] [CrossRef]
- Horigome, K.; Suzuki, D. Drying mechanism of Poly(N-isopropylacrylamide) microgel dispersions. Langmuir 2012, 28, 12962–12970. [Google Scholar] [CrossRef]
- Takizawa, T.; Sazuka, Y.; Horigome, K.; Sakurai, Y.; Matsui, S.; Minato, H.; Kureha, T.; Suzuki, D. Self-Organization of soft hydrogel microspheres during the evaporation of aqueous droplets. Langmuir 2018, 34, 4515–4525. [Google Scholar] [CrossRef]
- Mohanty, P.S.; Richtering, W. Structural ordering and phase behavior of charged microgels. J. Phys. Chem. B 2008, 112, 14692–14697. [Google Scholar] [CrossRef]
- Minami, S.; Yamamoto, A.; Oura, S.; Watanabe, T.; Suzuki, D.; Urayama, K. Criteria for colloidal gelation of thermo-sensitive poly(N-isopropylacrylamide) based microgels. J. Colloid Interface Sci. 2020, 568, 165–175. [Google Scholar] [CrossRef]
- Lopez-Leon, T.; Ortega-Vinuesa, J.L.; Bastos-Gonzalez, D. Cationic and anionic Poly(N-isopropylacrylamide) based submicron gel particles: Electrokinetic properties and colloidal stability. J. Phys. Chem. B 2006, 110, 4629–4636. [Google Scholar] [CrossRef]
- Sierra-Martin, A.; Romero-Cano, M.S.; Fernandez-Nieves, A.; Fernandez-Barbero, A. Thermal control over the electrophoresis of soft colloidal particles. Langmuir 2006, 22, 3586. [Google Scholar] [CrossRef]
- Ohshima, H.; Nakamura, M.; Kondo, T. Electrophoretic mobility of colloidal particles coated with a layer of adsorbed polymers. Colloid Polym. Sci. 1992, 270, 873–877. [Google Scholar] [CrossRef]
- Ohshima, H. Theory of electrostatics and electrokinetics of soft particles. Sci. Technol. Adv. Mater. 2009, 10, 063001. [Google Scholar] [CrossRef]
- Makino, K.; Yamamoto, S.; Fujimoto, K.; Kawaguchi, H.; Ohshima, H. Surface structure of latex particles covered with temperature-sensitive hydrogel layers. J. Colloid Interface Sci. 1994, 166, 251–258. [Google Scholar] [CrossRef]
- Daly, E.; Saunders, B. Temperature-Dependent electrophoretic mobility and hydrodynamic radius measurements of poly(N-isopropylacrylamide) microgel particles: Structural insights. Phys. Chem. Chem. Phys. 2000, 2, 387–3193. [Google Scholar] [CrossRef]
- Nabzar, L.; Duracher, D.; Elaissari, A.; Chauveteau, G.; Pichot, C. Electrokinetic Properties and Colloidal Stability of Cationic Amino-Containing N-Isopropylacrylamide−Styrene Copolymer Particles Bearing Different Shell Structures. Langmuir 1996, 14, 5062–5069. [Google Scholar] [CrossRef]
- Fernandez-Nieves, A.; Marquet, M. Electrophoresis of ionic microgel particles: From charged hard spheres to polyelectrolyte-like behavior. J. Chem. Phys. 2005, 122, 084702. [Google Scholar] [CrossRef] [PubMed]
- Hoare, T.; Pelton, R. Electrophoresis of functionalized microgels: Morphological insights. Polymer 2005, 46, 1139–1150. [Google Scholar] [CrossRef]
- Varga, I.; Kardos, A.; Borsos, A.; Gilanyi, T. Effect of internal charge distribution on the electrophoretic mobility of poly(N-isopropylacrylamide) based core-shell microgel particles. J. Mol. Liq. 2020, 302, 111979. [Google Scholar] [CrossRef]
- Maki, Y.; Sugawara, K.; Ngai, D. Temperature dependence of electrophoretic mobility and hydrodynamic radius of microgels of poly(N-isopropylacrylamide). Gels 2018, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Brugger, B.; Richtering, W. Emulsions stabilized by stimuli-sensitive Poly(N-isopropylacrylamide)-co-methacrylic acid polymers: Microgels versus low molecular weight polymers. Langmuir 2008, 24, 7769–7777. [Google Scholar] [CrossRef] [PubMed]
- Brugger, B.; Rosen, A.; Richtering, W. Microgels as stimuli-responsive stabilizers for emulsions. Langmuir 2008, 24, 12202–12208. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Zhang, Y.J.; Guan, Y.; Zhu, X.X. Gelation kinetics of thermosensitive PNIPAM microgel dispersions. Macromol. Chem. Phys. 2011, 212, 2052–2060. [Google Scholar] [CrossRef]
- Mansson, L.K.; De Wild, T.; Peng, F.; Holm, S.H.; Tegenfeldt, J.O.; Schrtberger, P. Preparation of colloidal molecules with temperature-tunable interactions from oppositely charged microgel spheres. Soft Matter 2019, 15, 8512–8524. [Google Scholar] [CrossRef]
- Xia, L.W.; Ju, X.J.; Liu, J.J.; Xie, R.; Chu, L.Y. Responsive hydrogels with poly(NIPAM-co-acrylic acid) colloidal spheres as building blocks. J. Colloid Interface Sci. 2010, 349, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Kim, J.W.; Hyun, D.C.; Jeong, U.Y.; Weitz, D.A. Regulating volume transitions of highly responsive hydrogel scaffolds by adjusting the network properties of microgel building block colloids. Langmuir 2010, 26, 3854–3859. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Panteli, P.A.; Patrickios, C.S. Complex hydrogels based on multiply interpenetrated polymer networks: Enhancement of mechanical properties via network multiplicity and monomer concentration. Macromolecules 2018, 51, 7533–7545. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takad, T.; Haraguchi, R. Nanocomposite gels by initiator-free photopolymerization: Role of plasma-treated clay in the synthesis and network formation. ACS Appl. Nano Mater. 2018, 1, 418–425. [Google Scholar] [CrossRef]
- Sugimura, A.; Asai, M.; Matsunaga, T.; Akag, Y.; Sakai, T.; Noguchi, H.; Shibayama, M. Mechanical properties of a polymer network of Tetra-PEG gel. Polym. J. 2013, 45, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Mayumi, K.; Tezuka, M.; Bando, A.; Ito, K. Mechanics of slide-ring gels: Novel entropic elasticity of a topological network formed by ring and string. Soft Matter 2018, 8, 8179–8183. [Google Scholar] [CrossRef]
- Kureha, T.; Minato, H.; Suzuki, D.; Urayama, K.; Shibayama, M. Concentration dependence of the dynamics of microgel suspensions investigated by dynamic light scattering. Soft Matter 2019, 15, 5390–5399. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawaguchi, H. On Going to a New Era of Microgel Exhibiting Volume Phase Transition. Gels 2020, 6, 26. https://doi.org/10.3390/gels6030026
Kawaguchi H. On Going to a New Era of Microgel Exhibiting Volume Phase Transition. Gels. 2020; 6(3):26. https://doi.org/10.3390/gels6030026
Chicago/Turabian StyleKawaguchi, Haruma. 2020. "On Going to a New Era of Microgel Exhibiting Volume Phase Transition" Gels 6, no. 3: 26. https://doi.org/10.3390/gels6030026
APA StyleKawaguchi, H. (2020). On Going to a New Era of Microgel Exhibiting Volume Phase Transition. Gels, 6(3), 26. https://doi.org/10.3390/gels6030026