4.2. Synthesis
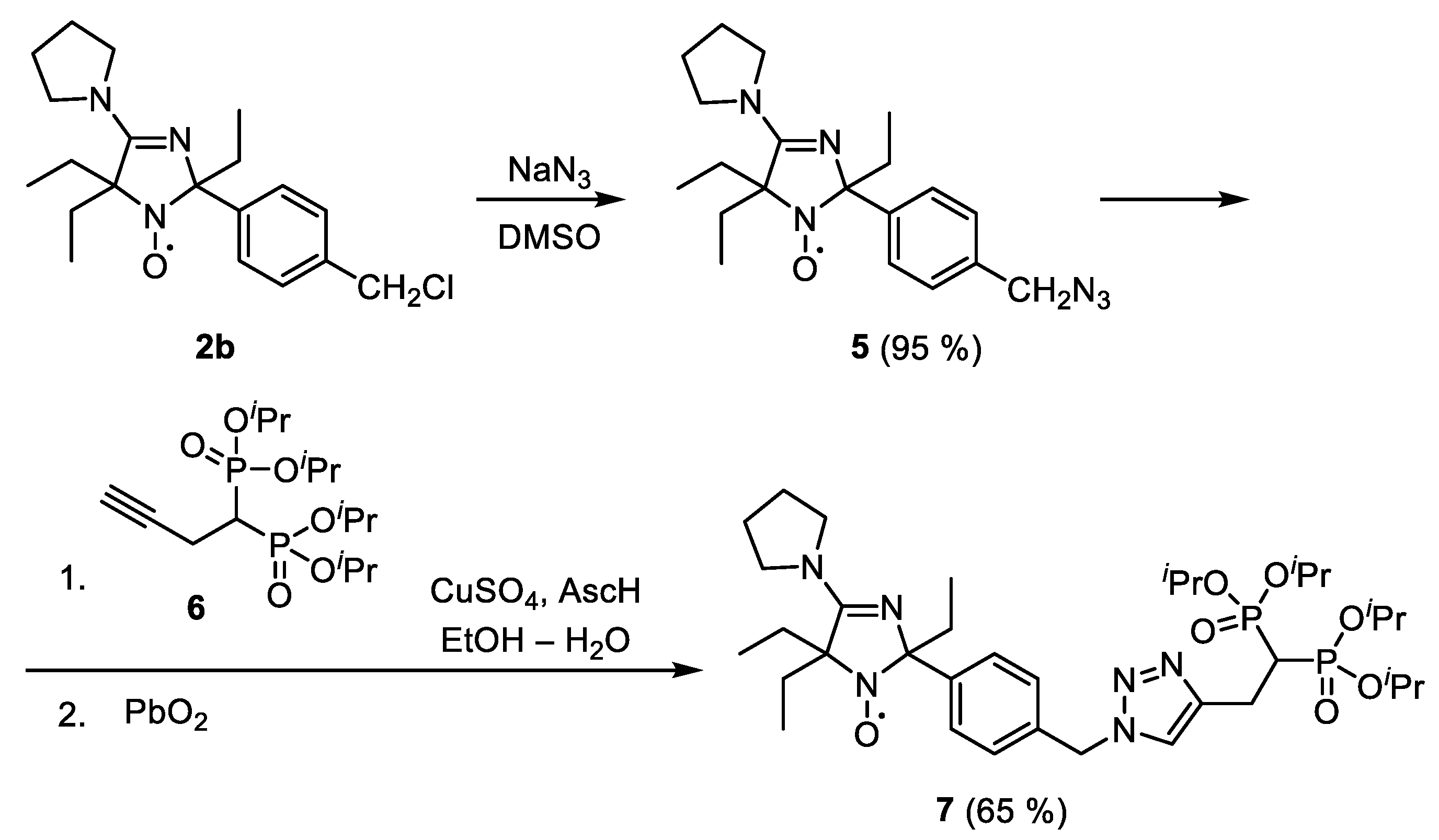
2-(4-(Azidomethyl)phenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (5)
A mixture of
2b hydrochloride [
15] (320 mg, 0.82 mmol), sodium hydrocarbonate (250 mg, 3 mmol), diethyl ether (30 mL), and water (20 mL) was vigorously stirred until powder of 2b completely dissolved. The ether solution was separated and concentrated in vacuum without heating. The residue was dissolved in DMSO (5 mL), a solution of NaN
3 (0.5 g, 7.7 mmol) was added, and the mixture was stirred at 60 °C for 10 h. The mixture was diluted with water (20 mL) and saturated solution of NaCl (50 mL) and extracted with diethyl ether. The extract was washed with saturated solution of NaCl and dried with Na
2CO
3, concentrated in vacuum, and the residue was separated using column chromatography on silica gel, eluent chloroform, to give
5, yield 290 mg (95%), yellow crystals, m.p. 63–65 °C (hexane). Elemental analysis, found: C, 65.33; H, 8.01; N, 22.47; calcd. for C
20H
29N
6O: C, 65.01; H, 7.91; N, 22.75%. IR (KBr) ν
max: 2091 (N
3), 1597 and 1574 (C=N, C=C).
Tetraisopropyl but-3-yne-1,1-diyldiphosphonate (6)
(In analogy to procedure by C. Li and C. Yuan [
41]) Tetraisopropyl methylenediphosphonate (5 g, 14.5 mmol) was added dropwise to a stirred suspension of NaH (0.8 g 50% content, 16.7 mmol) in dry THF (50 mL) under argon. After hydrogen evolution ceased, propargyl bromide (1.1 mL, 14.5 mmol) was added dropwise under argon to the stirred suspension. The mixture was stirred for 3 h, then the mixture was diluted with water (50 mL) and pH was adjusted to neutral with hydrochloric acid. The mixture was extracted with diethyl ether, the extract was dried with Na
2CO
3 and concentrated in vacuum. The residue was separated using column chromatography on silica gel, eluent chloroform, to give
6, yield 1.4 g (25%), colorless liquid. Elemental analysis, found: C, 50.35; H, 8.49; P, 16.10; calcd. for C
16H
32O
6P
2: C, 50.26; H, 8.44; P, 16.20%; IR (neat) ν
max (cm
−1): 2122 (C≡C).
1H NMR (400 MHz; CDCl
3,
δ): 1.25 (24H, m, CH
3), 1.93 (1H, t,
J 2.3, ≡CH), 2.43 (1H, br tt,
Jt1 24,
Jt2 5.9, P−CH−P), 2.64 (2H, tdd,
Jt 16,
Jd1 5.9,
Jd2 2.3, CH
2), 4.7 (4H, m, O−CH<);
13C{
1H} NMR (150 MHz; CDCl
3, δ): 15.87 (t,
JP 4.8, CH
2), 23.70 (dd,
JP-1 5.8,
JP-2 1.4, CH
3), 24.05 (t,
JP 3.5, CH
3), 37.87 (t,
JP 135.7, P
2CH), 69.66 (s, ≡CH), 71.36 (dd,
JP-1 5.7,
JP-2 6.5, OCH), 81.60 (t,
JP 9.7, –C≡).
2-(4-((4-(2,2-Bis(diisopropoxyphosphoryl)ethyl)-1H-1,2,3-triazol-1-yl)methyl)phenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (7)
Ascorbic acid (140 mg, 0.79 mmol) was added to a mixture of 5 (241 mg, 0.65 mmol), 6 (259 mg, 0.67 mmol), EtOH (1.5 mL), H2O (1.5 mL), and saturated solution of CuSO4 in water (0.15 mL). The mixture was stirred for 2 h, then PbO2 (1 g, 4.17 mmol) was added, the mixture was stirred for 1 h, then the precipitate was filtered off and washed with ethanol. The combined solutions were evaporated in vacuum and the residue was separated using column chromatography on silica gel, eluent chloroform, to give 7, yield 320 mg (65%), yellow oil. Elemental analysis, found: C, 57.23; H, 8.30; N, 10.98; P, 8.35; calcd. for C36H61N6O7P2: C, 57.51; H, 8.18; N, 11.18; P, 8.24%; IR (neat) νmax (cm−1): 1595, 1576 (C=N, C=C). 1H NMR (300 MHz; CD3OD–CDCl3, reduced with Zn in ND4Cl/D2O, δ): 0.76 (3H, t, J 7, CH3), 0.90 (3H, t, J 7, CH3), 1.04 (3H, t, J 7, CH3), 1.26 (24H, m, C(CH3)2), 1.45 (2H, m, CH2Me), 1.83 (1H, m, CH2Me), 2.12 (7H, br m, CH2Me and C-CH2CH2-C), 2.86 (1H (partly exchanged), tt, JP 23, JH 6, P–CH–P), 3.26 (2H, br t, JP 16.5, P2C−CH2−), 3.7 (4H, m, CH2−N−CH2), 4.69 (4H, septet, J 6, O−CH), 5.51 (2H, s, Ar−CH2), 7.32 (2H, d, J 8, CH Ar), 7.39 (1H, s, OH), 7.52 (2H, d, J 8, CH Ar), 7.69 (1H, s, N−CH=); 1H NMR (300 MHz; CD3OD–CDCl3, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 0.85 (3H, t, J 7, CH3), 0.89 (3H, t, J 7, CH3), 1.04 (3H, t, J 7, CH3), 1.19 (6H, d, J 6, C(CH3)2), 1.27 (6H, d, J 6, C(CH3)2), 1.33 (12H, d, J 6, C(CH3)2), 1.49 (2H, m, CH2Me), 2.02 (8H, br m, CH2Me and C−CH2CH2−C), 2.99 (1H (partly exchanged), tt, JP 24, JH 6, P−CH-P), 3.26 (2H, m, P2C−CH2−), 3.7 (4H, m, CH2−N−CH2), 4.75 (4H, septet, J 6, O−CH), 5.56 (2H, s, Ar−CH2), 7.37 (2H, d, J 8, CH Ar) and 7.60 (2H, d, J 8, CH Ar), 7.75 (1H, s, N−CH=); 31P NMR (121.497 MHz; CD3OD-CDCl3, reduced with Zn in ND4Cl/D2O, δ): 20.44, 20.47.
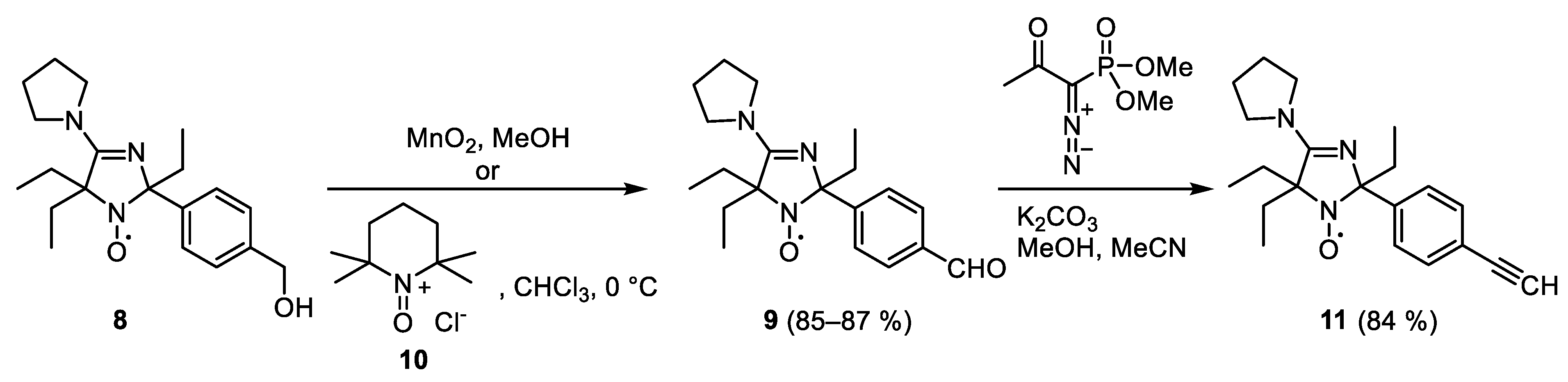
2,5,5-Triethyl-2-(4-formylphenyl)-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (9)
Method A
Activated manganese dioxide (4 g, 46 mmol) was added to a stirred solution of 8 (0.4 g, 1.16 mmol) in methanol (50 mL). The mixture was stirred for 4 h, manganese oxides were filtered off through celite 281, the solvent was distilled off in vacuum and the residue was separated using column chromatography on silica gel, eluent chloroform, to give 9, yield 340 mg (85%), yellow crystals, m.p. 84–85 °C dec. (chroloform-hexane). Elemental analysis, found: C, 70.20; H, 8.15; N, 12.31; calcd. for C20H28N3O2: C, 70.14; H, 8.24; N, 12.27%. IR (KBr) νmax (cm−1): 1697 (C=O); 1593, 1570 (C=N, C=C); UV (EtOH) λmax (log ε): 229 (4.22), 253 (4.26).
Method B
A powder of 2,2,6,6-tetramethylpiperidinium chloride (0.4 g, 2.03 mmol) was added to a solution of 8 (0.5 g, 1.5 mmol) in chloroform (10 mL) and the solution was stirred for 2 h at room temperature. The solvent was distilled off in vacuum, the residue was separated using column chromatography on silica gel, eluent chloroform, to give 9, yield 440 mg (85%).
2,5,5-Triethyl-2-(4-ethynylphenyl)-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (11)
A solution of dimethyl (1-diazo-2-oxopropyl)phosphonate in acetonitrile (10%, 0.7 mL, 0.31 mmol) was added to a mixture of 8 (100 mg, 0.29 mmol), freshly annealed K2CO3 (84 mg, 0.61 mmol) and anhydrous methanol (5 mL). The mixture was stirred overnight, methanol was distilled off in vacuum, the residue was triturated with ethyl acetate, the precipitate was filtered off and washed with ethyl acetate, the combined solution was concentrated in vacuum and separated by column chromatography on silica gel, eluent diethyl ether–hexane 1:1 to give 11, yield 82 mg (84%), orange crystals, m.p. 156–158 °C (hexane-ethyl acetate). Elemental analysis, found: C, 74.81; H, 7.97; N, 12.50; calcd. for C21H28N3O: C, 74.52; H, 8.34; N, 12.41%. IR (KBr) νmax (cm−1): 3151 (≡C–H), 2094 (C≡C); 1587, 1554 (C=N, C=C).
2-(4-Carboxyphenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (12)
A solution of 8 (0.5 g, 1.5 mmol) in CHCl3 (10 mL) was cooled to 0 °C and 2,2,6,6-tetramethyloxopiperidinium chloride (10) (0.4 g, 2.0 mmol) was added in one portion. The mixture was stirred for 2 h at 0 °C. Then 2-methylbut-2-ene (1.8 mL, 17.4 mmol) was added to reaction mixture followed by addition of a solution of NaClO2 (0.9 g, 9.8 mmol) and KH2PO4 (1.3 g, 9.8 mmol) in H2O (44 mL). The mixture was stirred for 2 h, the organic layer was separated, washed with saturated aqueous solution of Na2CO3 (3 × 20 mL) and concentrated in vacuum. The residue was separated by column chromatography on silica gel using CHCl3–EtOH mixture (100:16) as an eluent to give light-yellow crystals of 12, yield 463 mg (89%), m.p. 205–207 °C (AcOEt—i-PrOH 10:1). Elemental analysis, found: C, 66.85; H, 7.87; N, 11.71; calcd. for C20H28N3O3: C, 67.01; H, 7.87; N, 11.72%. IR (KBr) νmax (cm−1): 2974 (C–H), 1693 (C=O), 1591 (C=N), 1571 (C=C). UV (EtOH) λmax (log ε): 232 (4.41). 1H NMR (400 MHz; CD3OD–CDCl3, reduced with PhSH, δ): 0.78 (3H, t, J 7.3, CH3), 0.85 (3H, t, J 7.3, CH3), 0.95 (3H, t, J 7.3, CH3), 1.00–1.12 (2H, m, CH2, Et), 1.36, 1.75 (2H, AB, CH2, Et), 1.85–1.99 (2H, m, CH2, Et), 2.00 (4H, m, 4CH2, Pyrr), 3.50–3.55 (4H, m, CH2−N−CH2, Pyrr), 7.66 (2H, d, J 8, CH Ar), 8.01 (2H, d, J 8, CH Ar).
2-(4-(Ethoxycarbonyl)phenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (14)
Pyridine (340 μL, 4.2 mmol) was added to a suspension of 12 (0.5 g, 1.4 mmol) acid in dry CHCl3 (10 mL). The resulting solution was stirred at 0 °C, and SOCl2 (130 μL, 1.8 mmol) was added dropwise. The stirring continued for 3 h, then ethanol (1 mL, 17 mmol) was added in one portion. The mixture was stirred for 2 h, the solvent was removed in vacuum, and the residue was separated using column chromatography on silica gel, eluent CHCl3–EtOH 200:1, to give 14, yield 352 mg (65%), yellow crystals, m.p. 85–90 °C (hexane). Elemental analysis, found: C, 68.60; H, 8.10; N, 10.80; calcd. for C23H32N3O3: C, 68.27; H, 8.35; N, 10.87). IR (KBr) νmax (cm−1): 2970 (C–H), 1718 (C=O), 1593 (C=N), 1571 (C=C). UV (EtOH) λmax (log ε): 231 (4.45). 1H NMR (300 MHz; CDCl3-CD3OD, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 0.62 (3H, t, J 7.2, CH3), 0.68 (3H, t, J 7.4, CH3), 0.81 (3H, t, J 7.5, CH3), 0.88–1.12 (2H, m, CH2, Et), 1.17 (3H, t, J 7.1, CH3CH2O), 1.2, 1.60–1.84 (4H, m, CH2, Et2), 1.89 (4H, m, C−CH2CH2−C, Pyrr), 3.44 (4H, m, CH2–N–CH2, Pyrr), 4.15 (2H, q, J 7.1, CH2O), 7.44 (2H, d, J 8, CH Ar), 7.80 (2H, d, J 8, CH Ar).
2-(4-((2,5-Dioxopyrrolidinooxy)carbonyl)phenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (15)
Pyridine (100 μL, 1.2 mmol) was added to a suspension of 12 (0.138 g, 0.39 mmol) in dry CHCl3 (5 mL), the resulting solution was stirred at 0 °C, and SOCl2 (30 μL, 0.4 mmol) was added dropwise. The reaction mixture was stirred for 3 h, then N-hydroxysuccinimide (44 mg, 0.39 mmol) was added in one portion. The mixture was stirred for 1 h, the solvent was removed in vacuum, and residue was separated using column chromatography on silica gel, eluent CHCl3–EtOH (100:1), to give 15, yield 102 mg (58%), yellow crystals, m.p. 107–108 °C (Et2O—hexane 1:2). Elemental analysis, found: C, 63.09; H, 6.86; N, 11.92; calcd. for C24H31N4O5: C, 63.28; H, 6.86; N, 12.30%. IR (KBr) νmax (cm−1): 2974 (C–H), 1774, 1743 (C=O), 1591 (C=N), 1571 (C=C). UV (EtOH) λmax (log ε): 235 (4.41).
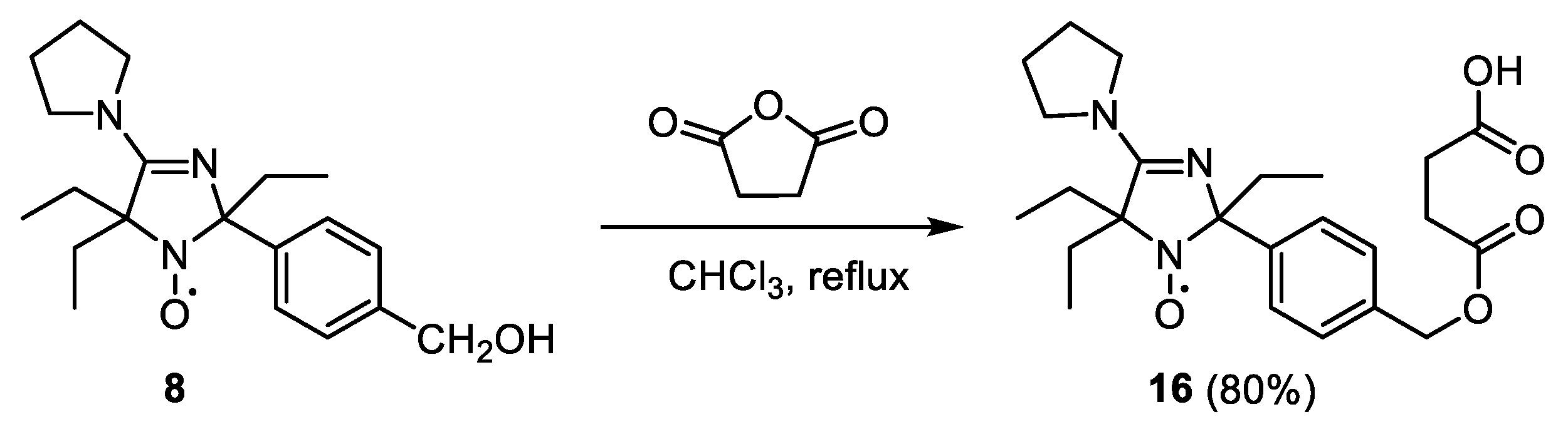
2-(4-((3-Carboxypropanoyloxy)methyl)phenyl)-2,5,5-triethyl-4-pyrrolidino-2,5-dihydro-1H-imidazol-1-oxyl (16)
Succinic anhydride (0.15 g, 1.5 mmol) was added to a solution of 8 (0.2 g, 0.6 mmol) in CHCl3 (10 mL) and the reaction mixture was heated to reflux for 2 h. The resulting solution was washed with H2O (10 mL), dried with Na2SO4, and the solvent was removed in vacuum. The solid residue was triturated with ether, the crystalline precipitate of 16 was filtered off and washed with diethyl ether, yield 213 mg (80%), yellow crystals, m.p. 168–169 °C dec. (Et2O). Elemental analysis, found: C, 64.37; H, 7.52; N, 9.48; calcd. for C24H34N3O5: C, 64.24; H, 7.71; N, 9.45%. IR (KBr) νmax (cm−1): 2969 (C-H), 1731 (C=O ester), 1587 (C=N), 1569 (C=O carboxy). UV (EtOH) λmax (log ε): 220 (4.03). 1H NMR (300 MHz; CDCl3-CD3OD, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 0.66 (6H, m, CH3), 0.81 (3H, m, CH3), 1.00, 1.24 (2H, m, CH2, Et), 1.56–1.81 (4H, m, 2 × CH2, Et2), 1.89 (4H, m, C−CH2CH2−C, Pyrr), 2.44 (4H, m, CH2CH2CO2H), 3.44 (4H, m, CH2–N–CH2, Pyrr), 4.9 (2H, m, CH2O), 7.14 (2H, d, J 8, CH Ar), 7.31 (2H, d, J 8, CH Ar).
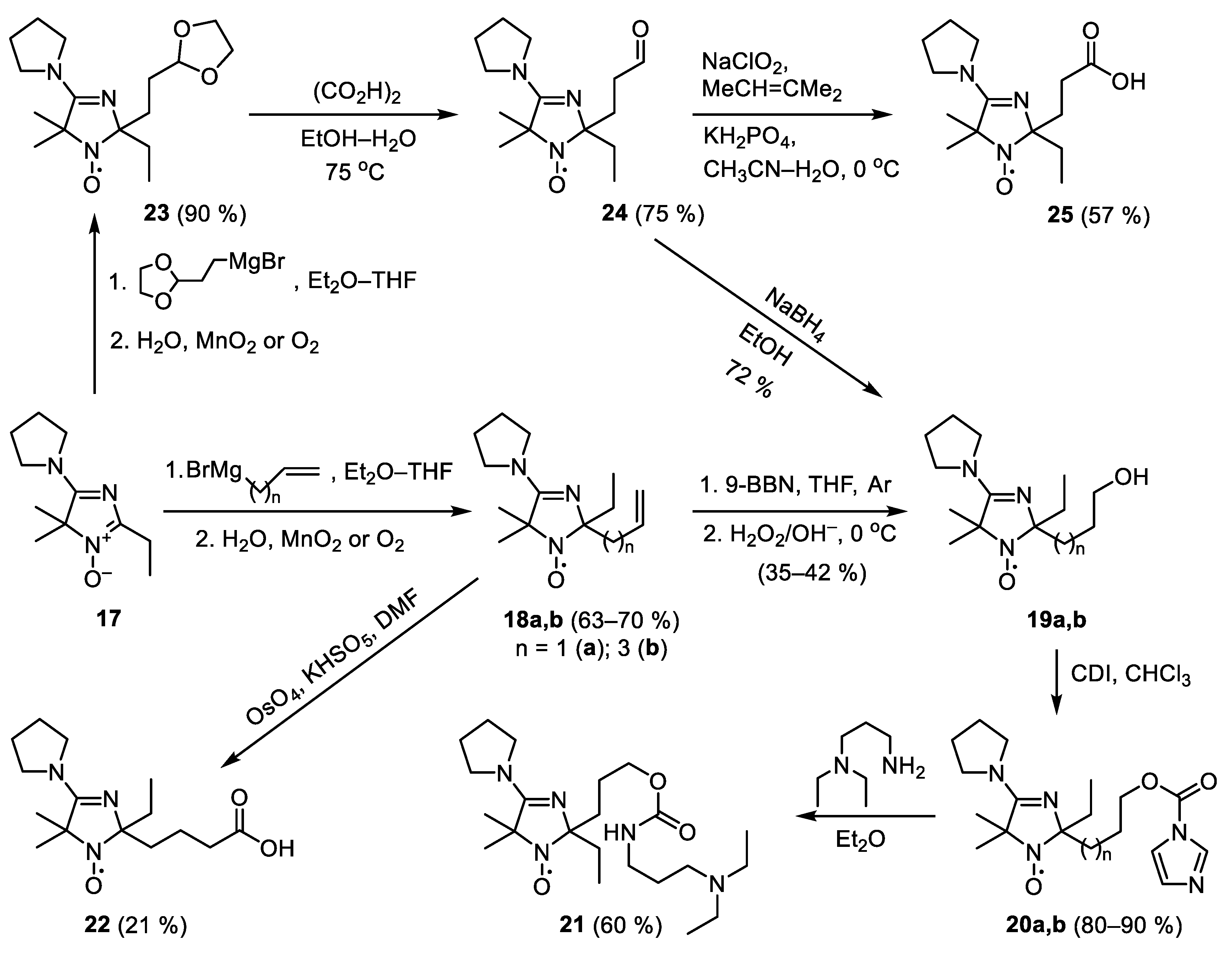
2-Allyl-2-ethyl-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydroimidazol-1-oxyl (18a)
A solution of allylmagnesium bromide prepared from allyl bromide (1.69 mL, 20 mmol) and Mg (0.5 g, 20.5 mmol) in diethyl ether (15 mL) under argon was added dropwise to a stirred solution of nitrone 17 (0.83 g, 4.0 mmol) in THF (15 mL). The reaction mixture was stirred for 1 h, then water (30 mL) was added dropwise under vigorous stirring. Then manganese dioxide (5 g, 57 mmol) was added and the reaction mixture was stirred for 1 h. The manganese oxides were filtered off and the precipitate was washed with tert-butylmethyl ether. The organic layer was separated, the water solution was saturated with NaCl and extracted with tert-butylmethyl ether. The combined organic extracts were concentrated in vacuum and the residue was separated using column chromatography on Al2O3, eluent tert-butylmethyl ether–hexane (1:1) to give 18a, yield 630 mg (63%), yellow crystals, m.p. 55–57 °C (hexane). Elemental analysis, found: C, 67.22; H, 10.23; N, 16.88; calcd. for C14H24N3O: C, 67.16; H, 9.66; N, 16.78%. IR (KBr) νmax (cm−1): 2975 (C–H), 1645 (C=C), 1590 (C=N). UV (EtOH) λmax (log ε): 225 (4.17). 1H NMR (400 MHz; CD3OD, reduced with N2D4, δ): 0.91 (3H, t, J 7, CH3), 1.47 (6H, d, J 2.2, CH3), 1.60–1.83 (4H, m, CH2, Et), 2.01 (4H, m, CH2–CH2 (pyrr)), 2.39–2.63 (2H, m, CH2–CH=CH2), 3.56 (4H, s, CH2–N–CH2), 5.08 (2H, m, CH2=CH), 5.97 (1H, tdd, Jt 7, Jd1 10.7, Jd2 17.2, CH2=CH).
2-Ethyl-5,5-dimethyl-2-(pent-4-enyl)-4-(pyrrolidino)-2,5-dihydroimidazol-1-oxyl (18b)
A solution of pent-4-enylmagnesium bromide was prepared from 5-bromopentene (1.6 g, 12 mmol) and Mg (335 mg, 14 mmol) in THF (20 mL) under argon. This solution was added dropwise to a stirred solution of 17 (1 g, 4.8 mmol) in THF (20 mL). The reaction mixture was stirred overnight, then water (4 mL) was added dropwise under vigorous stirring. The reaction mixture was vigorously stirred in air for 1 h, then organic layer was separated, and the aqueous layer was extracted with Et2O–EtOH (100:1). The combined organic extracts were dried with Na2SO4, solvents were distilled off in vacuum, and the residue was separated by column chromatography on Al2O3 using hexane–CHCl3 mixture (2:1) as an eluent to give 18b. Yield 931 mg (70%), yellow oil. Elemental analysis, found: C, 68.93; H, 9.80; N, 15.00; calcd. for C16H28N3O: C, 69.02; H, 10.14; N, 15.09%. IR (KBr) νmax (cm−1): 2973 (C–H), 1639 (C=C), 1594 (C=N). UV (EtOH) λmax (log ε): 225 (4.19). 1H NMR (300 MHz; CDCl3-CD3OD, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 0.65 (3H, m, CH3, Et), 1.16 (2H, m, CH2, Et), 1.23 (2H, s, CH3), 1.32 (4H, s, CH3), 1.36–1.64 (4H, m, CH2–CH2–Allyl), 1.77 (2H, m, CH2–C=), 1.85 (4H, m, CH2–CH2–CH2–CH2), 3.23, 3.43 (4H, m, CH2–N–CH2), 4.60–4.76 (2H, m, CH2=), 5.48 (1H, tdd, =CH–, Jt 7, Jd1 10.3, Jd2 17,1).
2-Ethyl-2-(3-hydroxypropyl)-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydroimidazol-1-oxyl (19a)
Method A
A solution of 9-BBN in THF (0.5 M, 8 mL, 4.1 mmol) was added dropwise to a stirred solution of 18a (400 mg, 1.6 mmol) in THF (10 mL) under argon. The reaction mixture was vigorously stirred for 4 h, then cooled to 0 °C and cold (0 °C) aqueous NaOH (20%, 10 mL) and cold (0 °C) H2O2 (30%, 3 mL) were added dropwise successively. The mixture was allowed to warm to room temperature upon stirring (ca. 2 h), organic layer was separated, dried with Na2CO3, and the solvent was distilled off in vacuum. The residue was dissolved in CHCl3 (25 mL), anhydrous Na2CO3 (1 g) was added, and mixture was allowed to stand overnight in air. The solution was concentrated in vacuum and separated by column chromatography on silica gel using CHCl3–EtOH mixture (100:4) as an eluent to give 19a. Yield 150 mg (35%), yellow oil. Elemental analysis, found: C, 62.53; H, 9.49; N, 15.45; calcd. for C14H26N3O2: C, 62.65; H, 9.76; N, 15.66%. IR (KBr) νmax (cm−1): 3386 (br., OH), 1592 (C=N). UV (EtOH) λmax (log ε): 225 (4.07). 1H NMR (400 MHz; CD3OD, reduced with N2D4, δ): 0.94 (3H, t, J 7.2, CH3, Et), 1.43 (6H, s, CH3), 1.53–1.92 (6H, m, CH2), 1.98 (4H, m, CH2-CH2-CH2-CH2), 3.52 (4H, s, CH2-N-CH2), 3.57 (2H, br. s, CH2O). 2-Ethyl-2-(5-hydroxypentyl)-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydroimidazol-1-oxyl (19b) was prepared similarly from 18b. Yield 42%, yellow crystals, m.p. 68–73 °C (Et2O). Elemental analysis found: C, 65.17; H, 10.56; N, 14.08; calcd. for C16H30N3O2: C, 64.83; H, 10.20; N, 14.18%. IR (KBr) νmax (cm−1): 3261 (br., OH), 1593 (C=N). UV (EtOH) λmax (log ε): 225 (4.1). 1H NMR (300 MHz; CDCl3–CD3OD, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 0.62 (3H, m, CH3, Et), 1.06 (4H, br. m, CH2–CH2–(CH2)2OH), 1.21–1.28 (8H, m, 2 × CH3, CH2, Et), 1.40 (2H, m, CH2–CH2OH), 1.57 (2H, m, >C(Et)–CH2), 1.79 (4H, br. m, C−CH2CH2−C, Pyrr), 3.20, 3.44 (4H, m, CH2–N–CH2, Pyrr), 3.26 (2H, t, J 6.5, CH2O).
Method B
Sodium borohydride (60 mg, 1.6 mmol) was added portionwise to a stirred solution of 24 (400 mg, 1.5 mmol) in EtOH (10 mL) at 0 °C. The reaction was controlled with TLC, Silufol UV-254, eluent CHCl3–EtOH (25:1). Inorganic residue was filtered off, the solution was distilled off in vacuum, and the residue separated by column chromatography as described above to give 19a. Yield 309 mg (72%).
2-(3-(1H-Imidazole-1-carbonyloxy)propyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydro-1H-imidazol-1-oxyl (20a)
Carbonyldiimidazole (80 mg, 0.49 mmol) was added to a solution of alcohol 19a (114 mg, 0.43 mmol) in dry CHCl3 (5 mL) and the mixture was allowed to stand for 24 h. The solution was washed with brine, dried with Na2SO4, and concentrated in vacuum. The residue was separated by column chromatography on silica gel using CHCl3–EtOH mixture (100:2) as an eluent, producing 20a as yellow oil. Yield 139 mg (90%). Elemental analysis, found: C, 59.69; H, 7.72; N, 19.45; calcd. for C18H28N5O3: C, 59.65; H, 7.79; N, 19.32%. IR (KBr) νmax (cm−1): 1760 (C=O), 1592 (C=N). UV (EtOH) λmax (log ε): 223 (4.17). 2-(5-(1H-Imidazole-1-carbonyloxy)pentyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydro-1H-imidazol-1-oxyl (20b) was prepared similarly, yield 80%, yellow oil. Elemental analysis, found: C, 61.30; H, 8.26; N, 17.70; cacld. for C20H32N5O3: C, 61.51; H, 8.26; N, 17.93%. IR (KBr) νmax (cm−1): 1762 (C=O), 1593 (C=N). UV (EtOH) λmax (log ε): 226 (3.99).
2-(3-(3-(Diethylamino)propylcarbamoyloxy)propyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydro-1H-imidazol-1-oxyl (21)
N,N-Diethyl-1,3-diaminopropane (50 mg, 0.38 mmol) was added to a solution of
20a (126 mg, 0.35 mmol) in dry Et
2O (5 mL), and mixture was allowed to stay for 24 h. The solution was concentrated in vacuum, and the residue was separated by column chromatography on Al
2O
3 using CHCl
3 as an eluent to give
21 (
Figure 3). Yield 82 mg (60%), yellow oil. Elemental analysis, found: C, 62.21; H, 10.01; N, 16.51; calcd. for C
22H
42N
5O
3: C, 62.23; H, 9.97; N, 16.49%. IR (KBr) ν
max (cm
−1): 1718 (C=O), 1593 (C=N). UV (EtOH) λ
max (log ε): 225 (4.19).
1H NMR (300 MHz; CDCl
3-CD
3OD, reduced with Zn/CF
3COOH in CD
3OD, 65 °C,
δ): 0.75 (3H, t,
J 7.2, CH
3), 1.11 (6H, t,
J 7.3, 2 × CH
3) 1.33 (6H, br., CH
3), 1.40 (6H, br., CH
3), 1.43–1.79 (8H, m, CH
3CH
2C,
1CH
2,
2CH
2,
5CH
2,), 1.90 (4H, br. m,
10CH
2,
11CH
2), 2.87–3.03 (8H, m,
4CH
2,
6CH
2,
7CH
2,
8CH
2), 3.36, 3.52 (4H, m,
9CH
2,
12CH
2), 3.84 (2H, m, CH
2O).
2-(3-Carboxypropyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidino)-2,5-dihydro-1H-imidazol-1-oxyl (22)
Osmium tetroxide (30 mg, 0.4 mmol) and oxone (1.77 g, 5.8 mmol) were added successively to a solution of 18b (400 mg, 1.4 mmol) in DMF (20 mL) and the mixture was stirred for 3 h. A powder of Na2SO3 (10 g, 63 mmol) was added in one portion. Inorganic precipitate was filtered off and washed with EtOH, the combined solution was evaporated to dryness in vacuum and the residue was separated by column chromatography on silica gel using EtOH as an eluent to give 22, yield 90 mg (21%), yellow oil. M+ (cacld./found) 296.1969/296.1972. IR (neat) νmax (cm−1): 2977 (C-H), 1664 (C=O), 1592 (C=N). λmax (EtOH)/nm: 225 (lgε 4.19).
2-(2-(1,3-Dioxolan-2-yl)ethyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidin-1-yl)-2,5-dihydro-1H-imidazol-1-oxyl (23)
A solution of 2-(1,3-dioxolan-2-yl)ethylmagnesium bromide was prepared from 2-(2-bromoethyl)-1,3-dioxolan (4.3 g, 24 mmol) and Mg (670 mg, 28 mmol) in 20 mL THF under a stream of argon. This solution was added dropwise to a stirred solution of nitrone 17 (850 mg, 4 mmol) in 20 mL Et2O and 6 mL THF. The reaction mixture was stirred overnight, then water (5 mL) was added dropwise under vigorous stirring. The reaction mixture was allowed to air for 1 h, then organic layer was separated, inorganic residue was quenched with Et2O–EtOH (100:1). An isolated organic layer was dried over Na2SO4, solvents were removed in vacuum. The residue was separated using column chromatography on Al2O3 using CHCl3 as an eluent to give 23, yield 1.13 g (90%), yellow oil. Elemental analysis, found: C, 62.08; H, 9.21; N, 13.43; calcd for C16H28N3O3: C, 61.91; H, 9.09; N, 13.54%. IR (KBr) νmax (cm−1): 2972 (C-H), 1593 (C=N), 1143 (C–O). λmax (EtOH)/nm: 225 (lgε 3.90).
2-Ethyl-5,5-dimethyl-2-(3-oxopropyl)-4-(pyrrolidin-1-yl)-2,5-dihydro-1H-imidazol-1-oxyl (24)
A solution of oxalic acid (180 mg, 2 mmol) in water (6 mL) was added to a solution of nitroxide 23 (250 mg, 0.8 mmol) in EtOH (4 mL). The reaction mixture was stirred for 3 h under reflux, then ethanol was removed in vacuum, saturated aqueous KHCO3 (10 mL) was added to a residue. The product was extracted with CHCl3–i-PrOH mixture (50:1) (3 × 15 mL). An isolated organic layer was dried over Na2SO4, the solvents were removed in vacuum, the residue was separated using column chromatography on silica gel using CHCl3-EtOH mixture (50:1) as an eluent to give 24, yield 161 mg (75%), yellow oil. Elemental analysis, found: %: C, 63.08; H, 9.18; N, 15.63; calcd. for C14H24N3O2: C, 63.13; H, 9.08; N, 15.78. IR (neat) νmax (cm−1): 2972 (C-H), 1720 (C=O), 1593 (C=N). λmax (EtOH)/nm: 225 (lgε 4.16).
2-(2-Carboxyethyl)-2-ethyl-5,5-dimethyl-4-(pyrrolidin-1-yl)-2,5-dihydro-1H-imidazol-1-oxyl (25)
Trimethylethylene (1 mL, 9.0 mmol) was added to a cooled (0 °C) solution of aldehyde 24 (200 mg, 0.8 mmol) in 10 mL CH3CN followed by addition of a solution of NaClO2 (480 mg, 5.3 mmol) and KH2PO4 (710 mg, 5.3 mmol) in H2O (20 mL). Progress of the reaction was monitored by TLC (silica gel, CHCl3–EtOH (50:1), developing with 1% aq KMnO4). CH3CN was removed in vacuum, the product was extracted from water by CHCl3–i-PrOH mixture (100:1) (5 × 15 mL). An isolated organic layer was dried over Na2SO4, the solvents were removed in vacuum, the residue was separated using column chromatography on silica gel using CHCl3–EtOH mixture (5:2) as an eluent to give 25, yield 121 mg (57%), yellow oil, M+ (calcd./found) 282.1812/282.1811. IR (neat) νmax (cm−1): 2973 (C–H), 1729 (C=O), 1591 (C=N). λmax (EtOH)/nm: 223 (lgε 4.04). 1H NMR (300 MHz; CDCl3-CD3OD, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ) 0.75 (3H, m, CH3, Et), 0.98–1.14 (2H, m, CH2, Et), 1.34, 1.40 (6H, m, 2 × CH3), 1.47–1.61 (2H, m, CH2CH2CO2H), 1.91 (4H, m, CH2–CH2–CH2–CH2, Pyrr), 2.14–2.27 (2H, m, CH2CH2CO2H), 3.36, 3.55 (4H, m, CH2–N–CH2, Pyrr).
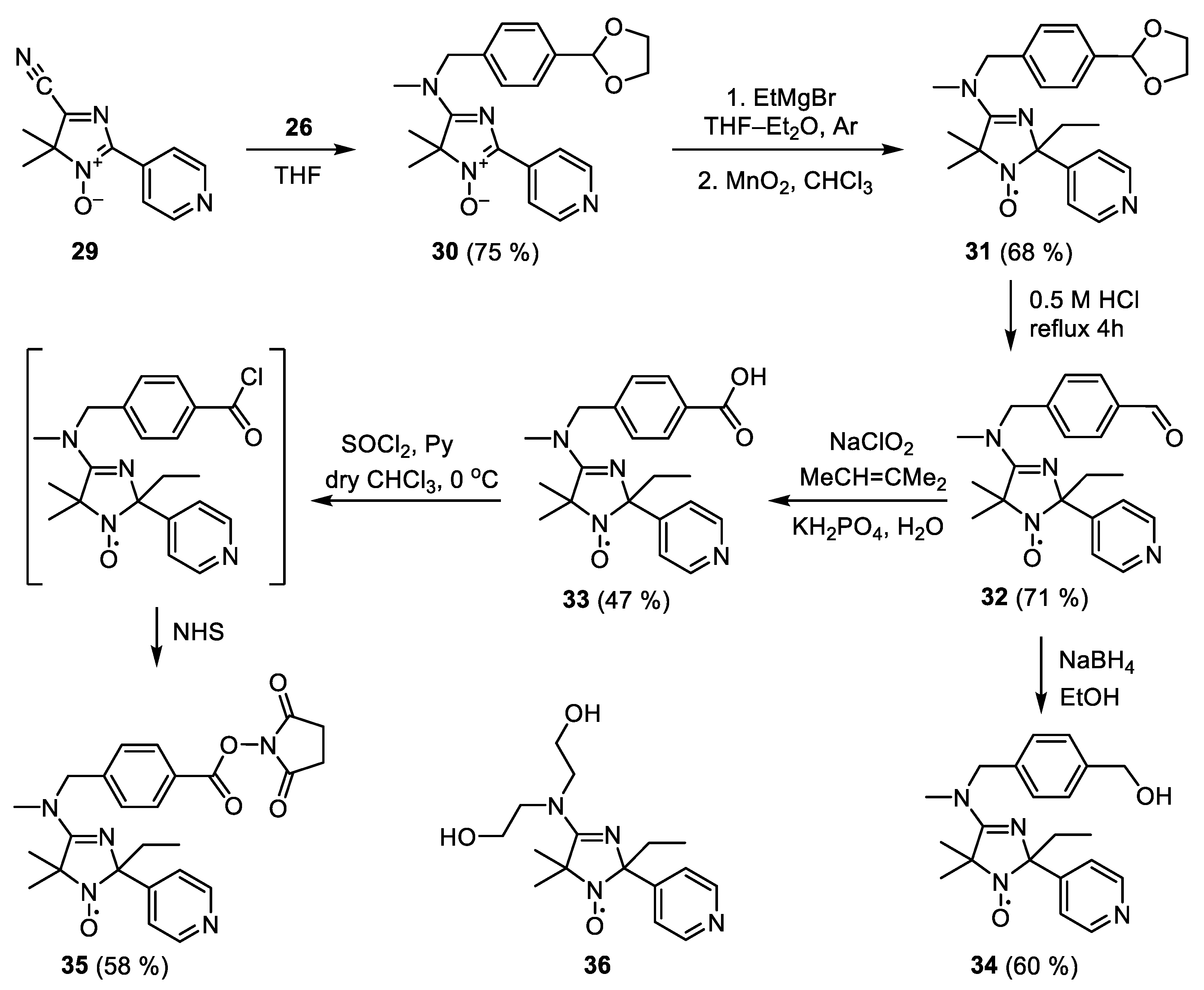
1-(4-(1,3-Dioxolan-2-yl)phenyl)-N-methylmethanamine (26)
p-Toluene sulfonic acid monohydrate (0.5 g, 74.6 mmol) was added to a solution of terephthalic aldehyde 27 (10 g, 74.6 mmol) in 175 mL PhCH3. Water was distilled off with Dean-Stark tube. The reaction mixture was then quenched with aqueous NaHCO3, dried over Na2CO3, the solvent was remove in vacuum, and residue was dissolved in methanol saturated with methylamine (20 mL). The resulting solution was added to the previously maintained under vigorous stirring for 10 min in a solution of Ti(Oi-Pr)4 (14 mL, 47 mmol) in methanol saturated with methylamine (30 mL). The mixture was stirred for 5 h, then NaBH4 (1.34 g, 33.6 mmol) was added portionwise, and mixture was stirred for 2 h. Water (7 mL) was added dropwise, solvents were removed in vacuum, brine was added to a residue, and the product was extracted by ether. Organic layer was dried over NaOH. Residue was separated using column chromatography on silica gel using Et2O–EtOH mixture (10:1) as an eluent, yield 12.24 g (85%), colorless oil. Elemental analysis, found: C, 67.84; H, 7.91; N, 6.94; calcd for C11H15NO2: C, 68.37; H, 7.82; N, 7.25%. 1H NMR (300 MHz; CDCl3, δ): 2.38 (3H, s, CH3), 3.70 (2H, s, N–CH2), 3.95–4.09 (4H, m, –O–CH2–CH2–O–), 5.75 (1H, s, O–CH–O), 7.29, 7.38 (4H, AA’BB’, C6H4)), 13C NMR (75 MHz; CDCl3, δ): 35.77 (N–CH3), 55.56 (N–CH2), 65.06 (O–CH2–CH2–O), 103.44 (O–CH–O), 126.31 (CH–C–CH2NHCH3), 127.89 (CH–C–CH), 136.32 (C–CH2NHCH3), 141.10 (C–CH). IR (neat) νmax (cm−1): 3325 (N–H), 1082 (O–C–O). λmax (EtOH)/nm: 210 (logε 3.94), 260 (logε 2.36).
5-((4-(1,3-Dioxolan-2-yl)benzyl)(methyl)amino)-4,4-dimethyl-2-(pyridin-4-yl)-4H-imidazole 3-oxide (30)
1-(4-(1,3-Dioxolan-2-yl)phenyl)-N-methylmethanamine 26 (6.72 g, 34.8 mmol) was added to a solution of 5-cyano-4,4-dimethyl-2-(pyridin-4-yl)-4H-imidazole 3-oxide 29 (2.98 g, 13.9 mmol) in THF (25 mL) and the mixture was allowed to stand at r.t. for 24 h. The solvent was removed in vacuum, residue was triturated with ether and crystallization from CH3CN to give 30, yield 3.97 g (75%), dirty-yellow crystals, m.p. 160 °C (dec.). Elemental analysis, found: C, 65.81; H, 6.25; N, 14.41; calcd for C21H24N4O3: C, 66.30; H, 6.36; N, 14.73%. 1H NMR (400 MHz; CDCl3, δ) 1.66 (6H, s, 2 × CH3), 3.06 (3H, s, N–CH3), 3.86–4.16 (4H, m, O–CH2–CH2–O), 4.77 (2H, br. s, N–CH2–Ar), 5.75 (1H, s, O–CH–O), 7.26, 7.46 (4H, AA’BB’, C6H4)), 8.46, 8.70 (4H, AA’BB’, Py). 13C NMR (75 MHz; CDCl3, δ) 21.50 (2 × Me), 35.22 (N–CH3), 53.11 (N–CH2), 65.00 (O–CH2–CH2–O), 75.73 (Me2C), 102.82 (O–CH–O), 121.00 (3,5–Py), 126.77 (br., CH (C6H4)), 133.86 (Py, i), 136.36 (C–CH2NCH3), 137.47 (C–CH), 144.67 (C=N→O), 149.82 (2,6-Py), 172.33 (C=N). IR (KBr) νmax (cm−1): 1597 (C=N), 1082 (O–C–O). λmax (EtOH)/nm: 263 (logε 4.30), 389 (logε 3.78).
4-((4-(1,3-Dioxolan-2-yl)benzyl)(methyl)amino)-2-ethyl-5,5-dimethyl-2-(pyridin-4-yl)-2,5-dihydro-1H-imidazol-1-oxyl (31)
A solution of ethylmagnesium bromide was prepared from ethyl bromide (2.73 g, 25 mmol) and Mg (630 mg, 26 mmol) in 35 mL Et2O under a stream of argon. This solution was added dropwise to a stirred solution of nitrone 27 (1 g, 2.6 mmol) in 15 mL THF. The reaction mixture was allowed to stand for 1 h. Then water (3 mL) was added dropwise under vigorous stirring followed by MnO2 (3 g, 34.5 mmol) addition. Progress of the reaction was monitored by TLC (silica gel, CHCl3–EtOH (100:3), developing with 1% aq. KMnO4). The mixture was stirred vigorously for 2 h, the oxidant was filtered off and the residue was washed by CHCl3 and MeOH. The solvent from filtrate was removed in vacuum and the residue was separated by column chromatography on silica gel using CHCl3-EtOH (100:3) as an eluent. The product 31 was isolated as a hydrochloride. Yield 797 mg (68%), yellow oil. Elemental analysis, found: C, 62.18; H, 6.83; N, 12.48; Cl, 6.70; calcd. for C23H30ClN4O3: C, 61.94; H, 6.78; N, 12.56; Cl, 6.95%. IR (neat) νmax (cm−1): 1593 (C=N), 1082 (O–C–O). λmax (EtOH)/nm: 216 (logε 4.34).
2-Ethyl-4-((4-formylbenzyl)(methyl)amino)-5,5-dimethyl-2-(pyridin-4-yl)-2,5-dihydro-1H-imidazol-1-oxyl (32)
A solution of nitroxide 30 (1.8 g, 4.4 mmol) in 15 mL 0.5 M aq. HCl was refluxed for 4 h, then Na2CO3 added to the end of gas evolution. A product was extracted by mixture of 20 mL CHCl3 + 1 mL i-PrOH three times, organic layer was dried over Na2CO3, the solvents were removed in vacuum, and the nitroxide 29 was isolated from the residue by column chromatography on silica gelusing CHCl3 as an eluent. Yield 1.14 g (71%), yellow oil. Elemental analysis, found: C, 68.73; H, 6.88; N, 14.92; calcd. for C21H25N4O2: C, 69.02; H, 6.90; N, 15.33%. IR (KBr) νmax (cm−1): 1701 (C=O), 1593 (C=N). λmax (EtOH)/nm: 252 (logε 4.29).
4-((4-Carboxybenzyl)(methyl)amino)-2-ethyl-5,5-dimethyl-2-(pyridin-4-yl)-2,5-dihydro-1H-imidazol-1-oxyl (33)
Trimethylethylene (1.33 g, 19.2 mmol) was added to a cooled (0 °C) solution of aldehyde 29 (583 mg, 1.6 mmol) in 20 mL CHCl3 followed by addition of a solution of NaClO2 (1.02 g, 11.2 mmol) and KH2PO4 (1.5 g, 11.2 mmol) in H2O (50 mL). Progress of the reaction was monitored by TLC (silica gel, CHCl3–EtOH (50:1), developing with 1% aq. KMnO4). The organic layer was separated, the product was extracted from water by CHCl3—i-PrOH mixture (20:1) (2 × 20 mL). A combined organic extracts were washed with brine, dried over Na2SO4, the solvents were removed in vacuum, the residue was separated using column chromatography on silica gel using AcOEt–EtOH mixture (10:1) as an eluent. Yield 285 mg (47%), yellow crystals, compound 33 was isolated as a crystal solvate 3 (33) × 2 EtOH (ether–EtOH 100:2), m.p. 204 °C (dec.). Elemental analysis, found: C, 65.12; H, 6.42; N, 13.12; calcd. for C67H87N12O11: C, 65.08; H, 7.09; N, 13.59%. IR (KBr) νmax (cm−1): 2474 (O–H), 1708 (C=O), 1597 (C=N), λmax (EtOH)/nm: 242 (logε 4.20).
2-Ethyl-4-((4-(hydroxymethyl)benzyl)(methyl)amino)-5,5-dimethyl-2-(pyridin-4-yl)-2,5-dihydro-1H-imidazol-1-oxyl (34)
NaBH4 (54 mg, 1.4 mmol) was added portionwise to a cooled (0 °C) solution of aldehyde 32 (511 mg, 1.4 mmol) in EtOH (20 mL). The reaction mixture was stirred until the reaction was complete (TLC, Silufol UV-254, eluent AcOEt). The solvent was removed in vacuum, the residue was separated using column chromatography on silica gel using AcOEt as an eluent. Yield 308 mg (60%), yellow crystals, compound 34 was isolated as a crystal solvate 2 (34) × 3 H2O (ether), m.p. 147–148 °C. Elemental analysis, found: C, 66.31; H, 7.12; N, 14.55; calcd. for C63H85N12O8: C, 66.47; H, 7.53; N, 14.76%. IR (KBr) νmax (cm−1): 3178 (O-H), 1595 (C=N). λmax (EtOH)/nm: 220 (logε 4.30). 1H NMR (400 MHz; CD3OD–CDCl3, reduced with Zn/CF3COOH in CD3OD, 65 °C, δ): 1.03 (3H, t, J 7.2, CH3 Et2), 1.29 (3H, br s, CH3), 1.79 (2H, q, J 7.2, CH2), 1.90 (3H, s, CH3), 3.20 (3H, br s, NCH3), 4.65 (2H, s CH2OH), 4.97 (2H, br s, N–CH2), 7.26 (2H, m, Ar), 7.46 (2H, m, Ar), 7.94 (2H, d, J 6.5, Py), 8.73 (2H, d, J 6.5, Py)
4-((4-(((2,5-Dioxopyrrolidin-1-yl)oxy)carbonyl)benzyl)(methyl)amino)-2-ethyl-5,5-dimethyl-2-(pyridin-4-yl)-2,5-dihydro-1H-imidazol-1-oxyl (35)
Pyridine (240 μL, 3 mmol) was added to a cooled (0 °C) suspension of acid 33 (228 mg, 0.6 mmol) in 10 mL of dry CHCl3 followed by addition of SOCl2 (90 μL, 1.2 mmol). The reaction mixture was vigorously stirred for 3 h, then N-hydroxysuccinimide (138 mg, 1.2 mmol) was added and the mixture was allowed to stand for 24 h. The solvents were then removed in vacuum, residue was separated using column chromatography on silica gel using CHCl3–EtOH mixture (100:2) as an eluent to give 35, yield 123 mg (40%), yellow crystals, compound 35 was isolated as a hydrochloride (hexane), m.p. 58 °C (dec.). Elemental analysis, found: C, 58.42; H, 5.47; N, 13.25; Cl, 6.56; calcd for C25H29ClN5O5: C, 58.31; H, 5.68; N, 13.60; Cl, 6.88%. IR (KBr) νmax (cm−1): 2976 (C-H), 1770 (O=C-N-C=O), 1741 (C=O), 1593 (C=N). λmax (EtOH)/nm: 239 (logε 4.25)


 and
and 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}