The polymer concentration should be higher than the overlap concentration
ϕ* to facilitate chain associations in the overlapping region to develop a physical gelation between a-PNIPAM chains.
ϕ* can be estimated by the inverse of the intrinsic viscosity, that is, ≈[
η]
−1.
Figure 1 shows the plot of
ηsp/
c versus
c at 20 °C, where
ηsp and
c are the specific viscosity and polymer concentration, respectively. Based on the Huggins equation
ηsp/
c = [
η]
+ k’[
η]
2 c, the extrapolated intercept at
c = 0 gives rise to the [
η] of 148.8 mL/g. Thus, the
ϕ* of the present a-PNIPAM in de-ionized water is approximately 0.6 wt%. The entanglement concentration
ϕe has been determined at 8 wt% [
14,
15]. In this work, a-PNIPAM solutions with various concentrations (5–12 wt%) were used to investigate their rheological properties, from which the corresponding
Ts was determined.
2.1. Rheological Curves Obtained from Temperature Sweep Test
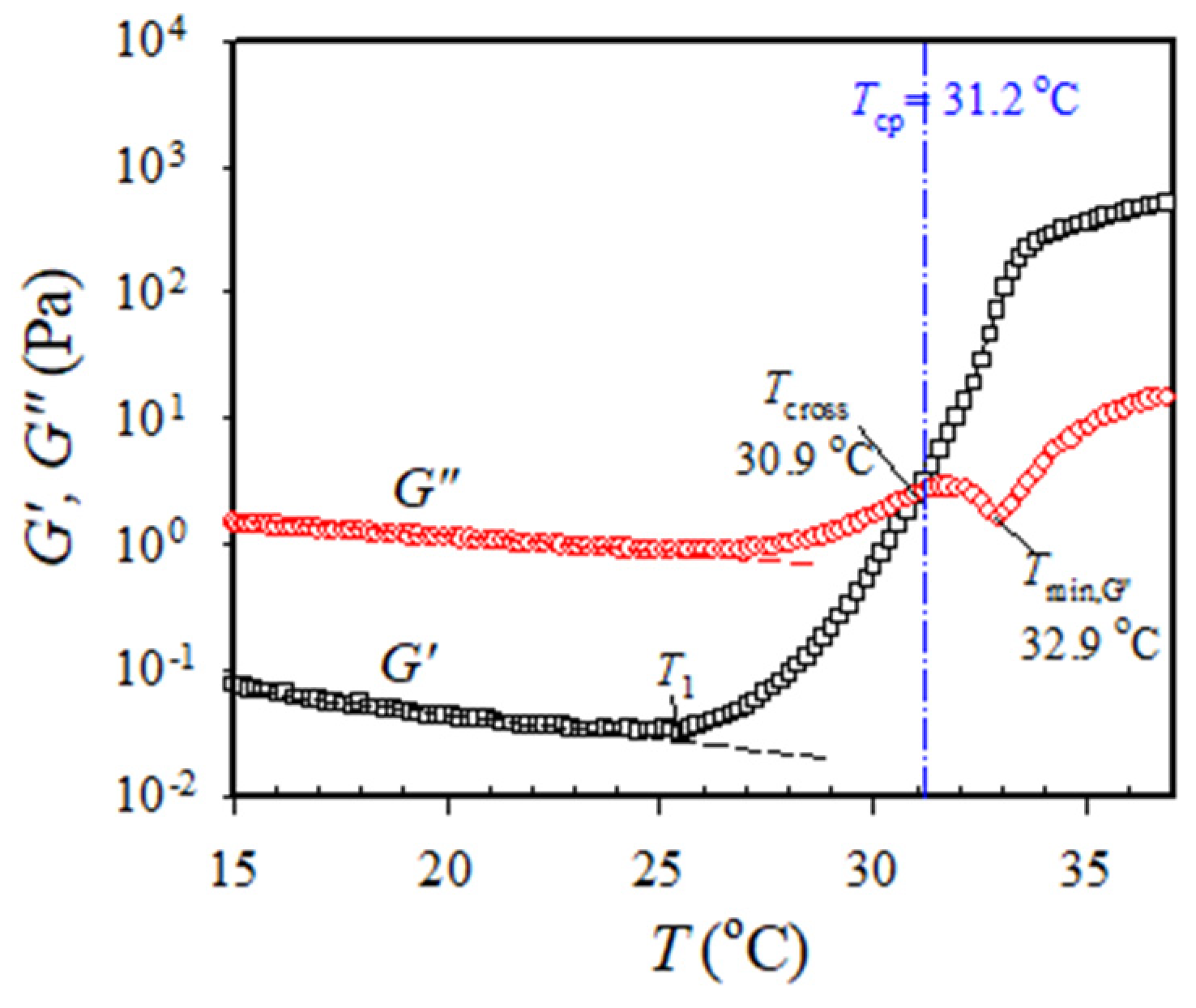
Figure 2 shows the typical
G′ and
G″ variation obtained from the temperature sweep test for the 7 wt% a-PNIPAM solution subjected to an applied frequency of 5 rad/s at a heating rate of 0.2 °C/min. The linear viscoelastic response was ascertained under a strain amplitude of 10% at the temperature range of 10–40 °C. Upon heating from 10 °C,
G′ and
G″ gradually decreased with increasing temperature until 25–26 °C, at which an upturn is observed. The upturn of
G′ happens slightly earlier than the
G″; the corresponding temperature is designated by
T1. At the temperature range of 10–25 °C,
G″ is one order magnitude higher than
G′, thereby inferring that viscous nature is dominant over elastic nature. There is an expected continuous decrease of
G′ and
G″ with
T which is attributed to the enhanced chain mobility at high temperatures. The upturn of
G′ and
G″ curves with
T indicates that the statistical mechanical characteristics of the solution in the single phase have been altered as follows. At
T >
T1, the solution starts to have such enhanced concentration fluctuations that must fulfill the criterion of their characteristic relaxation times being larger than the time scale of the observation specified by 1/
ω. Such thermal concentration fluctuations are developed at temperatures much below the cloud point of 31.2 °C determined from the turbidity at 0.2 °C/min heating as shown by the vertical dash–dot line.
The onset temperature at which
G′ upturns with
T indicates the onset of the enhanced concentration fluctuations to develop the branched chains by chain association [
14], thereby, implying that the one-phase solution at
T <
T1 eventually reaches the pre-gel regime at the temperature of
T1. Remarkably, a continuous and significant increase in
G′ with a three-order magnitude enhancement at the temperature range of 25–33 °C was observed. The increase in
G′ becomes mild above 33 °C. In contrast, the
G″ curve at the temperature range of 25–33 °C exhibits distinct temperature dependence, whereby an evident
G″ minimum is seen at 32.9 °C (denoted by
Tmin,G”), immediately followed by a second
G″ uprise. More importantly, a crossover of the
G′ versus
T curve and
G″ versus
T curve is detected at 30.9 °C, above which
G′ becomes dominant over
G″. The crossover temperature is denoted by
Tcross, thereby inferring the occurrence of macroscopic physical gelation. However, it should be reminded that
Tcross is not the gel point (GP at
Tgel) given that the critical relaxation exponent (
n) for the present a-PNIPAM/H
2O system is 0.758, regardless of the a-PNIPAM concentration [
14]. It is reminded that
Tcross can be strictly taken as
Tgel only under the condition of a stoichiometrically balanced end-linking network with
n = 0.5 [
25].
Similar trends are observed for the 12 wt% solutions (
Figure 3a), whereby both the
T1 and
Tcross are lowered to 22.0 °C and 29.0 °C, respectively; however, the
Tmin,G” remains relatively unchanged at 32.8 °C. In addition, the
G′-G″ crossover temperature is 1.4 °C lower than the cloud point which is 30.4 °C. The magnitude of
tan δ significantly decreases from 4.0 in the one-phase solution at
T <
T1 to 0.03 as the physical gel becomes phase-separated at
T >
Tcp, as shown in
Figure 3c. A distinct minimum of
tan δ could also be observed at
Tmin,G”. For the crosslinking network with
n ≠ 0.5, a convenient approach proposed by Winter [
26] is applied to extract the
Tgel by plotting the normalized dynamic moduli with a known value of
n, that is,
G″/sin(
nπ/2) and
G′/cos(
nπ/2) as a function of
T, from which the crossover temperature is the
Tgel. Such a plot is shown in
Figure 3b, and the derived
Tgel is 25.8 °C, which is consistent with the value of 25.0 °C obtained rigorously from the frequency-independence of
tan δ analysis [
14,
15]. Thus, the
Tcross obtained from the temperature sweep test is higher than the
Tgel for the present a-PNIPAM/H
2O solution, thereby suggesting that macroscopic gel has been already formed at
T =
Tcross before reaching the
Tcp.
The values of
G′ at
Tcross are 2 and 18 Pa for the 7 and 12 wt% solutions, respectively. These values are approximately two orders of magnitude lower than the modulus of conventional hydrogels of ~1 kPa. Thus, a very soft a-PNIPAM gel is developed because of its extremely low “cross-linking” density, being 2.9 × 10
−3 and 4.9 × 10
−2 mol/m
3 for the 7 and 12 wt% solutions, respectively [
14]. The apparent molecular weight between two physical junctions is ca. 3 × 10
6 − 2.5 × 10
8 g/mol, thereby suggesting that at least 4–37 primitive a-PNIPAM chains are intermolecularly associated with one another to develop the single strand of the gel network. The shapes of a-PNIPAM gels may effectively change freely because of the low cross-linking density [
25].
Each strand of the soft gel may be built up by dimers and trimers of single chains as its primary building blocks. The building blocks are further linked together into a long strand. Hence, the strand is composed of various single chains and is expected to be flexible. The dimers and trimers are composed of four and seven sub-chains in the case when the end-to-end associations of the single chains are less probable. The strands, the sub-chains in the strands, dimers, and trimers (or multimers in general) coexisting free from the strands, and single unassociated chains (unimers) may all have Gaussian-chain characteristics and indistinguishably contribute to the thermal concentration fluctuations. These unique features may indicate that upon heating, a-PNIPAM/water solutions undergo profound concentration fluctuations above T1. The lifetime of the fluctuations must be larger than the time scale of the G′ measurements, that is, 1/ω~0.2 s. The enhanced thermal concentration fluctuations further trigger the physical gelation process to develop the soft gel before phase separation at T > Tcp.
The upturn of
G′ is a general phenomenon that also occurs for polymer mixtures: the upturn may be attributed to (1) the thermal concentration fluctuations for the mixtures when the temperature approaches the spinodal point of the mixtures [
20], and (2) the existence of domain interface because of the phase separation via nucleation/growth for the off-critical mixtures when crossing the binodal point [
22]. Han et al. [
22] considered the obtained
T1 as the “apparent” binodal temperature and reported that
T1 is elevated at a higher heating rate for the off-critical blends based on the extensive studies of the binary blends of polybutadiene/polyisoprene (PB/PI). In their case, the thermal concentration fluctuations become large only very near but below the binodal point in the single-phase mixture or above the binodal point wherein the phase separation starts to occur. In addition, the physical origin of
T1 observed by them in their study on the PB/PI blends is quite different from the present a-PNIPAM/water solutions which form the macroscopic gel at a temperature lower than the cloud point as seen in
Figure 2 and
Figure 3.
The formation of physical gel is unambiguously confirmed by the large hysteresis loop, as shown in
Figure 4, where the temperature dependence of
G′ for the 12 wt% solution subjected to 0.2 °C/min heating to 32 °C, immediately followed by 0.2 °C/min cooling. Both the heating and cooling curves fail to coincide, and a hysteresis loop is observed, thereby indicating that the rheological behavior in the cooling process is quite different from that in the heating process. The hysteresis loop observed is primarily caused by the imbalance of rate between the interchain association and dissociation of the hydrophobic N-isopropyl groups at different temperatures. These interchain associations eventually form the 3D gel junctions when the solution temperature is higher than
Tgel (~25.8 °C). The gel junctions of the a-PNIPAM hydrogel primarily originated from the hydrophobic association between N-isopropyl groups [
3,
9]. “Hydrogen-bond breaking” between the water and amide group may further enhance the gel formation and its strength at elevated temperatures where cooperative dehydration occurs [
9,
10]. The pearl-necklace conformation of PNIPAM chains driven by the cooperative hydration implies that not all monomer units are hydrated along the backbone chain [
10]. Moreover, the strength of hydrophobic hydration (i.e., the hydration of apolar entities of N-isopropyl) depends on the configuration or tacticity of the amphiphilic PNIPAM chains [
12].
The magnitude of
G′ at 5 °C right after cooling is higher than that at 5 °C before heating, as seen in
Figure 4. Thus, a long holding time at 5 °C is required to homogenize the cooled solution via interchain dissociation. The long relaxation time is because of the slow diffusion of water into the hydrophobic clusters (or gel junctions). Similar hysteresis effects have been observed on the melting enthalpy of PNIPAM aqueous solution measured by DSC heating/cooling trace [
27,
28]. In addition, the recovery time is much longer than the chemical gels of PNIPAM [
28]. The difference in the hysteresis behavior between the chemical gels and physical gels of PNIPAM is likely related to the interchain associations and dissociations of the 3D network. The interchain associations in the chemical gels occur between the network strands. Thus, the associations involve a large cost of the elastic free energy of the network chains, which causes the associations less probable. On the contrary, in the physical gels, the interchain associations occur not only between the network strands but also between the network strands and the unimers or multimers, which are free from the strands. This makes the associations in the physical gels more extensive and intensive. Notably, a 3D transparent gel can be obtained until
Tcp at temperatures higher than
Tgel, above which a turbid gel is expected.
The temperature dependence of
G′ and
G″ curves (
Figure 2 and
Figure 3) may be altered if the solutions are subjected to different heating rates and applied frequencies. Thus, the effects of heating rate and applied frequency should be thoroughly investigated to extract meaningful material functions of
G′ and
G″ for further analyses. In general, the heating rate for the system must be as low as possible and not too far from thermal equilibrium. Moreover, a low frequency is desirable to trace the evolution of large-scale structures with a long relaxation time.
2.2. Effects of Heating Rate on the Rheological Curves
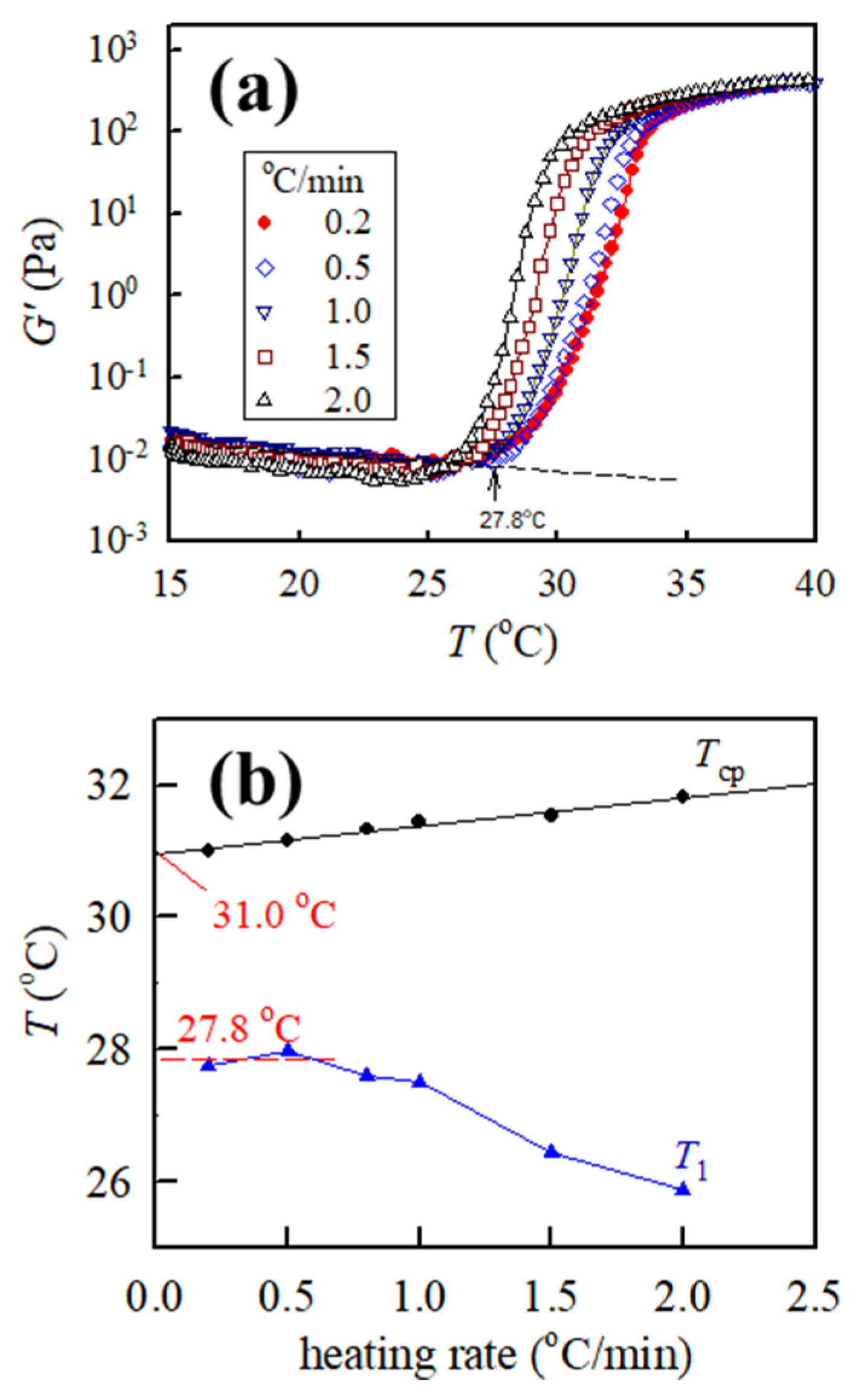
The 5.5 wt% solution was studied to reveal the heating rates on the obtained
G′ and
G″ curves; the corresponding curves at a heating rate of 0.2 °C/min and 2.0 °C/min are illustrated in
Figure 5a,b, with the same frequency of 5 rad/s, respectively. The most evident difference is the measured
G″ curves with the disappearance of the
Tmin,G” at a heating rate of 2.0 °C/min. Moreover, both
Tcross and
T1 decrease with increasing heating rates. These results indicate that heating rate plays an important effect on obtained rheological properties and selection of an appropriate heating rate is crucial.
Figure 6a shows the curves of
G′ versus
T for the 5.5 wt% a-PNIPAM solution obtained from different heating rates with the same frequency of 5 rad/s and strain amplitude of 10%. The 0.5 °C/min heating curve is marginally shifted to the low-temperature side, and the low-temperature shift becomes more pronounced at a higher heating rate (1.0 °C/min, 1.5 °C/min, and 2.0 °C/min) as compared with the heating curve at 0.2 °C/min. However, this trend is contrary to the turbidity measurement [
14,
15], thereby exhibiting that transmittance curves shift to the high-temperature side with an increased heating rate. Meanwhile,
Figure 6b shows the plots of
T1 and
Tcp versus heating rate. The extrapolated value of
Tcp at 0 heating rate is 31.0 °C, which is slightly lower than that determined from 0.2 °C/min heating (~31.4 °C). By contrast,
T1 remains unchanged at ~27.8 °C at low heating rates of 0.2 °C/min and 0.5 °C/min but is gradually decreased with a further increase in heating rate. The drop of
T1 at a heating rate higher than 0.5 °C/min is an artifact caused by the temperature measurement in the cup-and-bob fixture used in this study.
The measured
Tcp is relevant to the phase separation via nucleation/growth, which requires a certain induction time to proceed. Hence, the measured
Tcp depends upon the heating rate, whereby a higher heating rate delays the incipient indication in the nucleation/growth process up to a higher temperature. On the contrary, the determined
T1 is likely relevant to the onset of remarkable concentration fluctuations when the solution approaches the spinodal point [
20]. This criterion is particularly fulfilled for the critical solution, thereby exhibiting the so-called “critical slowing down” and “critical divergence” close to the critical point. The critical composition (
ϕc) for the a-PNIPAM/H
2O solution is ~16 wt% [
3]. Based on this conclusion, solutions with concentrations of 5–12 wt% studied in this work are off-critical. Even in this case, the contribution of thermal concentration fluctuations to enhance
G’ and
G” may still play an important role in general in the single-phase solutions below
Tcp (or
Tgel). Consequently,
T1 should be independent of the heating rate applied. Indeed, we found that
T1 remains relatively unchanged with the heating rate when it is lower than 0.5 °C/min. It seems that a heating rate of 0.2 °C/min used in this study is sufficient to extract the transition temperatures of
T1 and
Tcp. A complex structure evolution is involved in the a-PNIPAM aqueous solutions in the temperature range from
T1 to
Tcp.
2.5. Determination of Spinodal Temperature Based on the Fredrickson–Larson–Ajji–Choplin Mean Field Theory
Fredrickson and Larson [
32] developed a theoretical background based on the mean-field theory to disclose the contribution of the critical concentration fluctuations to the
G′ and
G″ in block copolymers during the order-disorder transition. They presented the contribution of the critical fluctuations to the linear viscoelastic of
G′(
ω,
T) and
G″(
ω,
T) based on the static structure factor
S0(
q;T) and the Onsager kinetics coefficient
λ(
q;T) for the two-component polymer systems. Their theoretical predictions validated the rheological measurements by Bates [
33], who showed the anomalous viscoelastic behavior of block copolymer in the pre-transitional regime from the one-phase disorder to the two-phase order state. After introducing the random phase approximation for the static structure factor, Ajji and Choplin [
20] applied the Fredrickson–Larson theory to the two-component polymer blends in the single-phase melt near critical point where the fluctuation contributions to
G′(
ω,
T) and
G″(
ω,
T) are anticipated to be dominant.
S0(
q;T) and
λ(
q;T) for the blends in the small
q-range are given by
where
and
Here,
ϕ is the volume fraction of the component 1;
Rg,K and
NK (
K = 1, 2) are the radius of gyration and the DP (degree of polymerization) of the
K-th component, respectively;
aK and
WK (
K = 1, 2) are the segmental length and the rate of segmental reorientation of the
K-th component, respectively.
F1 is a function of static parameters, whereas
F2 and
F3 depend on the dynamic parameters
WK which depend on
T. Equations (1) to (5) lead to
G′(
ω,
T) and
G″(
ω,
T) given by
where
kB is the Boltzmann constant. It is stressed that the quality
G″(
ω,
T)
2/
G′(
ω,
T) depends only on
F1, the statics of the concentration fluctuations but not on
F2 and
F3, the dynamics. Combined Equations (6) and (7) lead to
The temperature dependence of interaction parameter
χ is generally expressed as
χ =
A +
B/
T, where
A and
B are constants.
B is negative for the present system with LCST phase separation. By substituting
χ in Equation (8), the following equation is obtained:
where
α = (
kB/30
π)
2/3 and
Ts is the spinodal temperature. Based on Equation (9), the plot of (
G”2/
G′T)
2/3 versus 1/
T enables to access
Ts, provided that the concentration fluctuation contribution to
G′ and
G” are dominant. In Equation (9), the term
F1 is constant for a given polymer solution with no associating chains since
NK and
Rg,K are fixed, as defined in Equation (3). Thus, a linear dependence of (
G”2/G’T)
2/3 versus 1/
T is expected near the spinodal temperature in the single-phase solution, and
Ts can be derived by extrapolating the linear dependence to (
G”2/G’T)
2/3 = 0 and finding the intercept value (1/
Ts) with 1/
T axis without knowing the exact values of
B and
F1. Successful application of the Fredrickson–Larson–Ajji–Choplin (FLAC) theory to determine the
Ts for polymer blends [
20,
22,
23,
24] and polymer solution [
21] has been reported, and the derived
Ts is in fair agreement with that obtained from the scattering approach.
However, for the present gelling solution, in which a-PNIPAM chains are able to associate with one another to develop the multimers for the gel network, the application of Equation (9) is not straightforward and requires verification. In the associating systems, unimers (single unassociated chains) as well as dimers, rimers, …, and, etc., of a-PNIPAM chains coexist; their mole fractions may change with
T in such a way that the fraction of unimers tends to decrease but that of multimers to increase with increasing
T. The existence of the multimers certainly affects the function of
F1 to vary since the DP and chain configuration are changed as the solution temperature is raised. In this case, the structure factor for the associating chains is discussed in the Appendix in Ref. [
14], and the function of
F1 is derived to be
where
and
are the average square of the radius of gyration and the average DP of the associated chains in the gelling solution at
T. The definitions of
and
are given in Equations (11) and (12), respectively [
14].
In Equations (10)–(12), m is the association number of the chains and ϕm and are the concentration and mean square radius of gyration of the m-mer, respectively. The summation of ϕms with m gives the total polymer content in the solution, i.e., .
The magnitude of F1 may not be constant because the number of associating chains changes with T, thereby giving rise to the T dependence of and . The aqueous solutions of a-PNIPAM develop the macroscopic gel network at T= Tgel below Tcp, suggesting that the number of associating chains to form a m-mer increases with T to a large number of mgel at Tgel and that a further increase of m with T at Tgel < T < Tcp may be small compared with mgel. Under this condition, F1 is effectively independent of T at Tgel < T < Tcp. Accordingly, measured (G”2/G’T)2/3 as a function of T (>Tgel) enables to assess Ts, specifically from a perspective that the interchain associations strongly enhance the fluctuations, thereby giving rise to the extremely dominant contribution to G’ and G”.
Based on Equation (9), the rheological properties of the 7 and 12 wt% solutions are further analyzed, and the corresponding plots are provided in
Figure 9a. In addition, the respective values of
T1,
Tgel, and
Tcp are included to reveal the evolution of the (
G”2/G’T)
2/3 curves as the one-phase solution reaches the pre-gel regime (
T >
T1), transparent gel regime (
Tgel <
T <
Tcp), and opaque gel regime (
T >
Tcp), respectively. For the 7 wt% solution, a linear regression line is feasibly obtained at a first glance at temperatures higher than 25.8 °C (=
T1). However, a careful examination of the curve reveals that the linear slope at 1/
Tcp < 1/
T < 1/
T1 is not constant. The deviation in the linear slope in different temperature ranges is because of the possible interchain associations, thereby leading to the fact that the parameter
F1 is not constant but changes with temperature. The phenomenon is more evident for the curve of the 12 wt% solution.
Hence,
Ts,gel is the
Ts evaluated by extrapolating the plot using the data at
T >
Tgel for the macroscopic gel to account for this deviation. The final linear slope at 1/
Tcp < 1/
T < 1/
Tgel is extracted to derive a meaningful spinodal temperature of the a-PNIPAM gel, as shown by the blue solid line in
Figure 9b; the extrapolated intercept gives rise to
Ts,gel of 32.6 °C for the 7 wt% solution. For the 12 wt% solution, linear regression is deduced at temperatures above 28.2 °C (ca. 6 °C higher than the
T1 of 22 °C) to derive the
Ts,gel of 32.9 °C. Interestingly, the derived
Ts,gel is rather close to the experimentally observed
Tmin,G” regardless of solution concentrations (
Table 1), thereby implying a close correlation between the
Tmin,G” and the derived
Ts,gel. An abrupt increase in
G′ is seen at temperatures above
Tmin,G” (
Figure 2 and
Figure 3). The second uprise of
G′ and
G″ is attributed to the sudden formation of bicontinuous phase-separated domains associated with spinodal decomposition (SD). For the dynamically asymmetric solution of PNIPAN/water in a deep quench, the growth of the “3D percolated domain network” [
34] composed of the gel network will further enhance the solution elasticity with raising
T.
The correlation length
ξ of polymer solutions associated with the concentration fluctuations is increased when the solution temperature gradually approaches the spinodal point. The temperature dependence of
ξ can be expressed as [
20,
21]
For a given solution, the temperature dependence of
ξ can be calculated from the corresponding
G’ and
G”. Theoretically,
ξ diverges at
Ts.
Figure 10 shows the plots of (
G′T/
G″2)
1/3 versus
T for the solutions studied. The increase in
ξ is observed at temperatures above 30 °C, and the value of
ξ becomes maximum at
Tmin,G”. These plots confirmed that regardless of solution concentrations (5–12 wt%), all the solutions exhibit a similar
Ts,gel at 32–33 °C (shaded area in
Figure 10).
We have performed a detailed analysis of SAXS of the 12 wt% a-PNIPAM aqueous solution to extract the
Ts,gel after subtracting the excess scattering caused by the gel junctions from the observed scattering profile [
14]. In this manner, the deduced
Ts,gel by SAXS is 31.3 °C, which is higher than the
Ts of a-PNIPAM solution before physical gelation (~29.3 °C). The
Ts,gel derived from rheometry is higher than that obtained from the static scattering approach as compared to the results obtained from both approaches. Our determined values of
Ts,gel are further compared with those reported in the literature without addressing the issue of physical gelation.
Ts of PNIPAM aqueous solutions are determined from the Cahn linearized theorem to be 32.0–33.2 °C for the 4.4–7.3 wt% solutions based on the light scattering results [
19]. Shibayama et al. [
18] applied SANS to the 5.6–21.3 wt% PNIPAM/D
2O solutions, yielding
Ts values in the range of 33.1–35.1 °C. These results also show that
Ts is relatively independent of the PNIPAM concentration. The LCST phase diagram of PNIPAM/H
2O exhibits a bottom square-like shape, and this peculiar phase boundary has been theoretically validated by Okada and Tanaka [
10] through the cooperative hydration hypothesis.
2.6. Phase Diagram of a-PNIPAM/water Based on Rheometry and Turbidity
For the so-called “off-critical” a-PNIPAM/H
2O solutions of 5–12 wt% studied based on Ref. [
3] (
ϕc ~16 wt%), concentration fluctuations play a key role in the structure evolution when
Ts is approached below
Tb. The thermally-induced concentration fluctuations may occur over a wide range of length scales from ~nm to ~μm. The magnitude and length scale of concentration fluctuations both diverge at
Ts. Thus, we propose the following phase transitions on heating the a-PNIPAM/H
2O solution based on our rheological results.
At temperatures higher than T1, the concentration fluctuations in the single-phase solution become effective to yield interchain associations. However, the small length-scale fluctuation has only a small amplitude and rapidly decays. It barely facilitates intersegmental contacts and interchain associations. This is because a small-scaled region (~nm) rich in chain segments is stochastically hardly probable and thereby insignificant from the viewpoint of statistical mechanics. On the contrary, the large length-scale fluctuation has a very large amplitude of fluctuation and very slowly decays. It creates local regions where the local concentration of polymers is much higher than the average concentration. Moreover, these regions survive for a long time. These physical factors stochastically greatly facilitate the diffusion-controlled interchain associations mediated by attractive interactions between the hydrophobic groups to form the multimers. The rate of hydrophobic association is higher than the rate of hydrophobic dissociation, thereby giving rise to the formation of the associations.
The association mediated by the concentration fluctuations is diffusion-controlled in the regions rich in polymers where the local concentration is conceivably much higher than ϕe. The small multimers (e.g., dimers and trimers) built up by intermolecularly associated single chains act as gel junctions and primary building blocks of the network strands which eventually build up the macroscopic physical gel network at Tgel. The transparent gel turns opaque as the temperature is raised to Tb. When the temperature is further raised to Ts,gel or equivalently to Tmin,G”, the opaque solution confined in the soft gel undergoes a large-scale (~μm) viscoelastic phase separation.
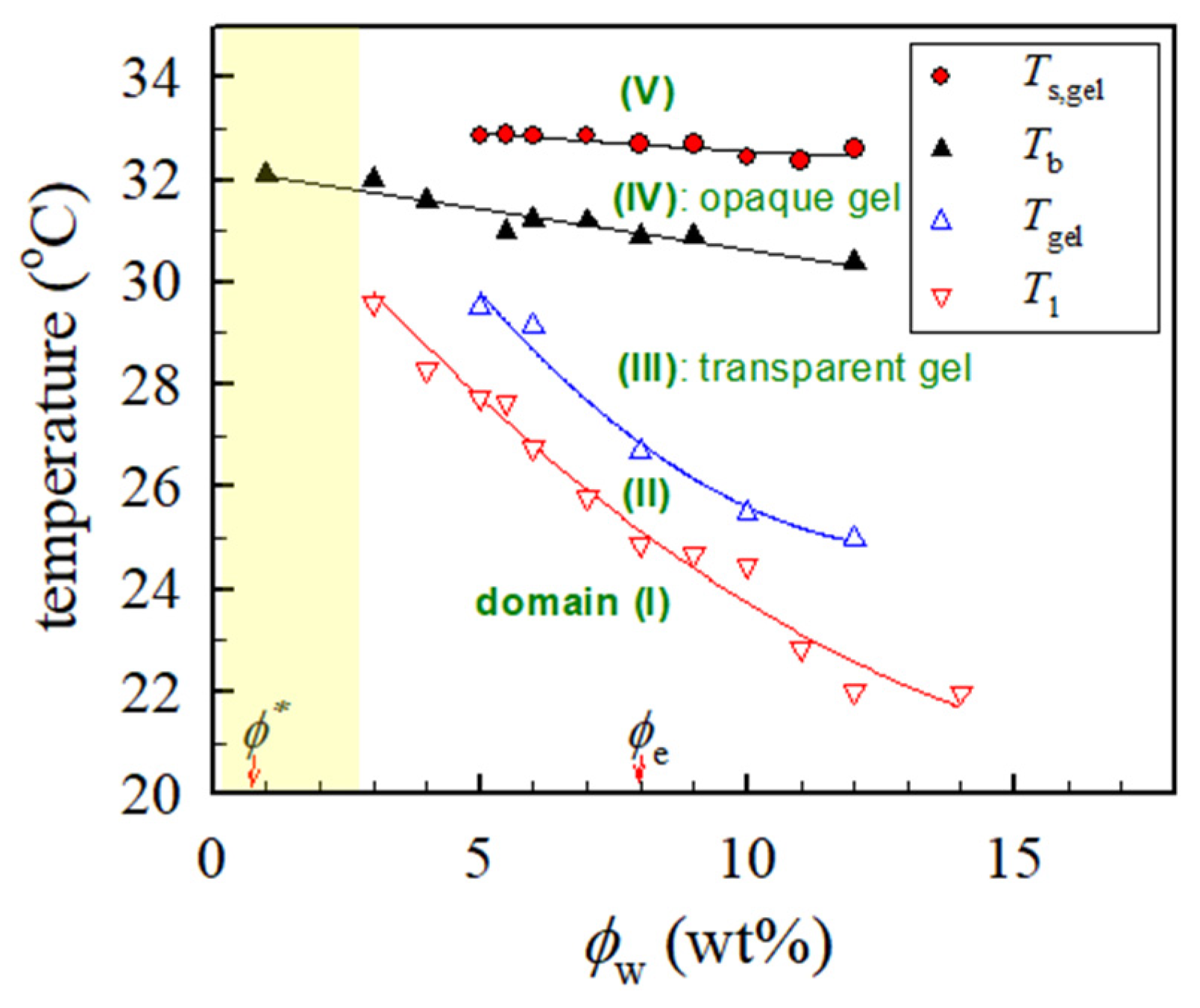
Figure 11 shows the phase diagram of a-PNIPAM aqueous solutions assessed by the linear viscoelasticity and turbidity. The corresponding values of
T1,
Tgel,
Tb, and
Ts,gel are included. However, the rheological properties of solutions at concentrations <3 wt% were not measured because of apparatus limitations; thus, the related data are absent, as represented by the shaded area. Five solution domains are identified in the quasi-equilibrium phase diagram depending upon the composition and temperature, namely, domains (I):
T <
T1, stable single-phase solution with the thermorheological simplicity, (II):
T1 <
T <
Tgel, the single-phase solution containing the interchain association subjected to profound concentration fluctuations, (III):
Tgel <
T <
Tb, transparent gel, (IV):
Tb <
T <
Ts,gel, opaque gel, and (V):
T >
Ts,gel, unstable gel undergoing viscoelastic phase separation via SD. The demarcation was primarily based on the dynamic temperature sweep test. A more advanced work of static scattering experiments is required to clarify the details of the structural evolution of the developing a-PNIPAM gels to extract the
Ts,gel to construct the “thermodynamic” phase diagram.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}