
sRNAflow: A Tool for the Analysis of Small RNA-Seq Data


Abstract
:1. Introduction
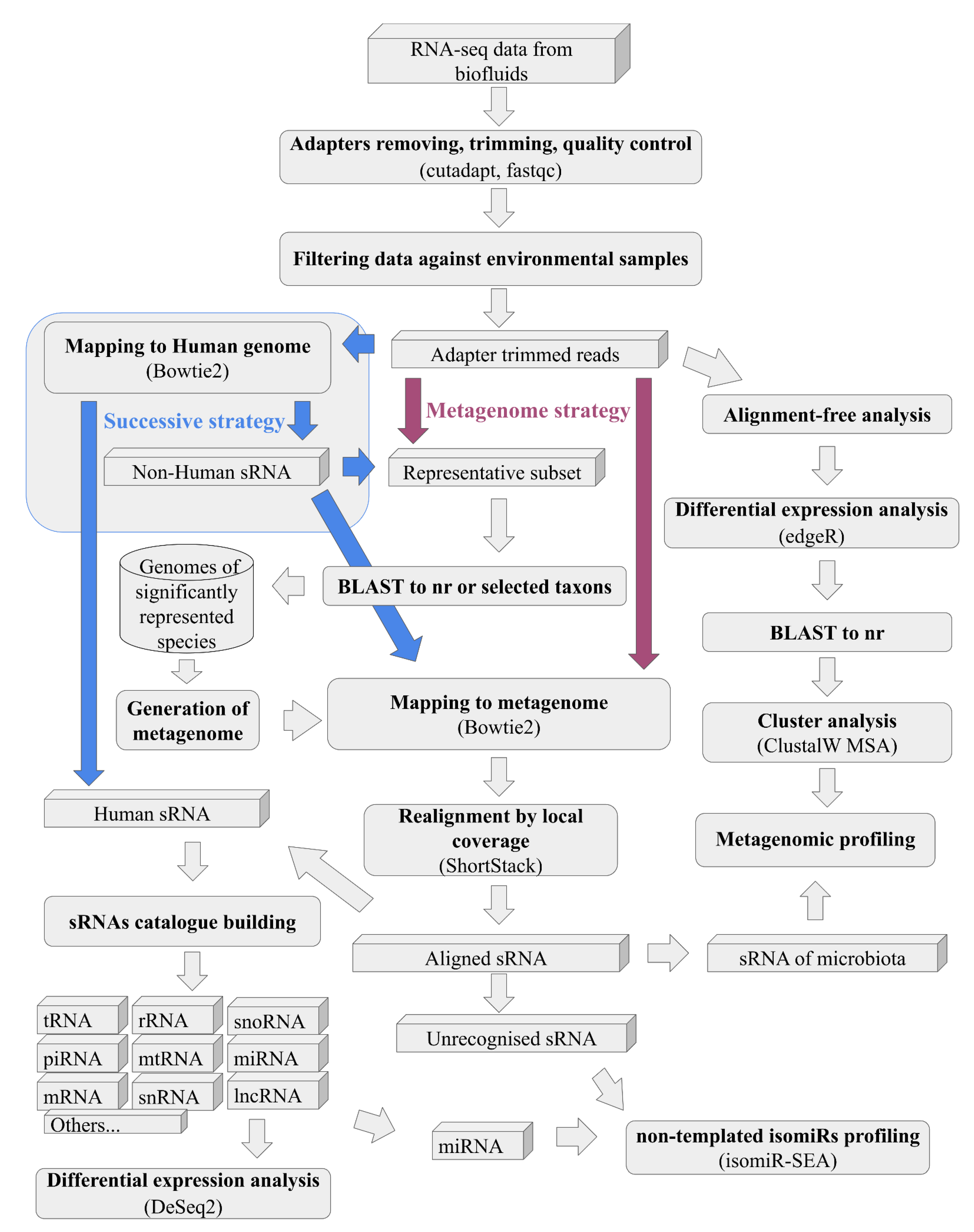
2. Materials and Methods
- Adapter removal and quality trimming (cutadapt [32]);
- BLAST of a representative subset of reads (BLAST [35]);
- Realignment by local coverage (ShortStack [22]);
- Reads counting (Rsubread [38]);
- Non-template isomiRs identification (isomiR-SEA [41]);
- Cluster analysis (ClustalW MSA [42]);
- Data visualisation (Krona [43]).
2.1. Installation
- mkdir -m 777 sRNAflow
- docker pull ghcr.io/zajakin/srnaflow
- docker run -d -p 3838:3838 -v `pwd`/sRNAflow:/srv/shiny-server/www ghcr.io/zajakin/srnaflow
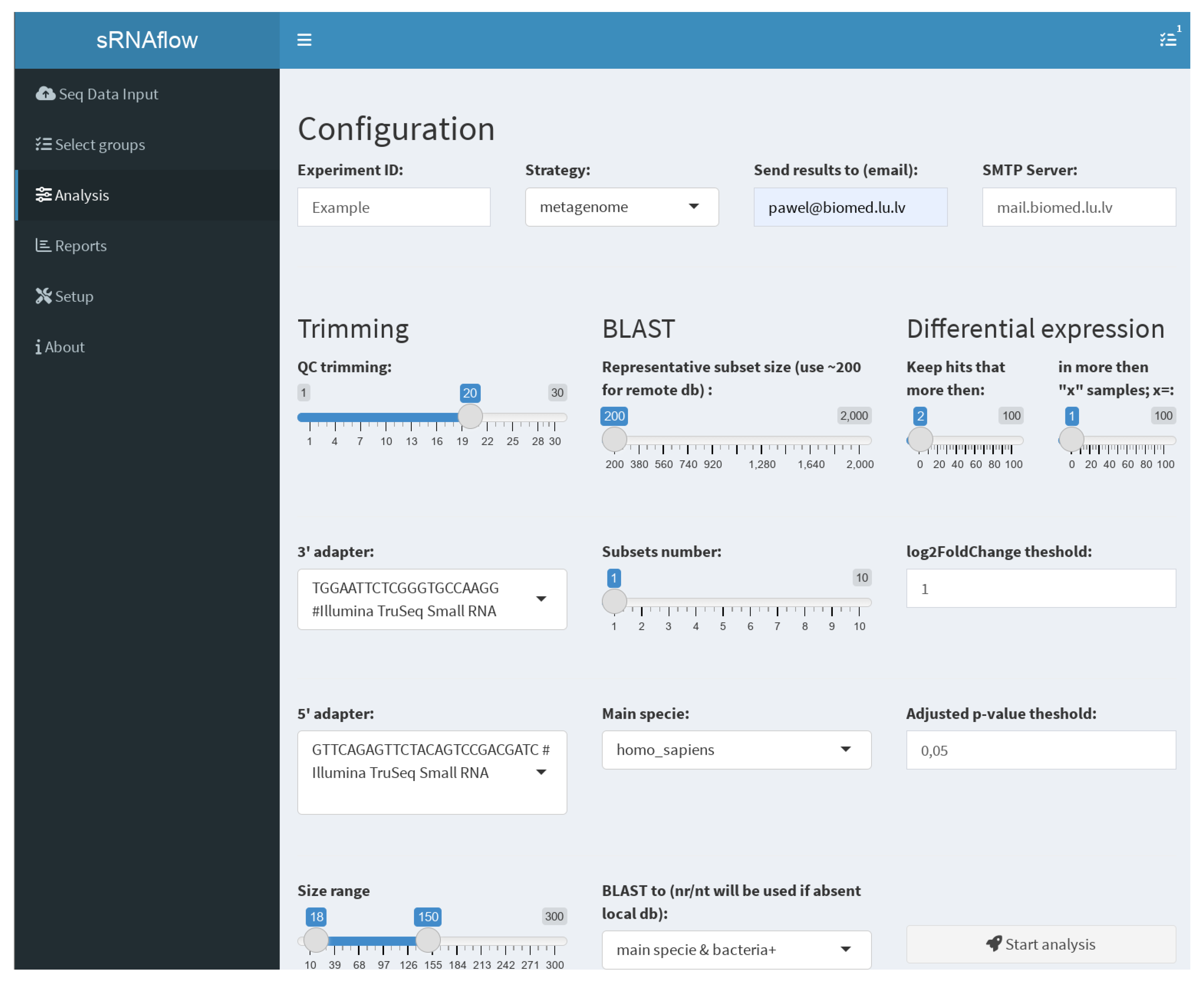
2.2. Shiny-Based User Interface
2.3. Annotations Files
2.3.1. Generation of Annotation Files
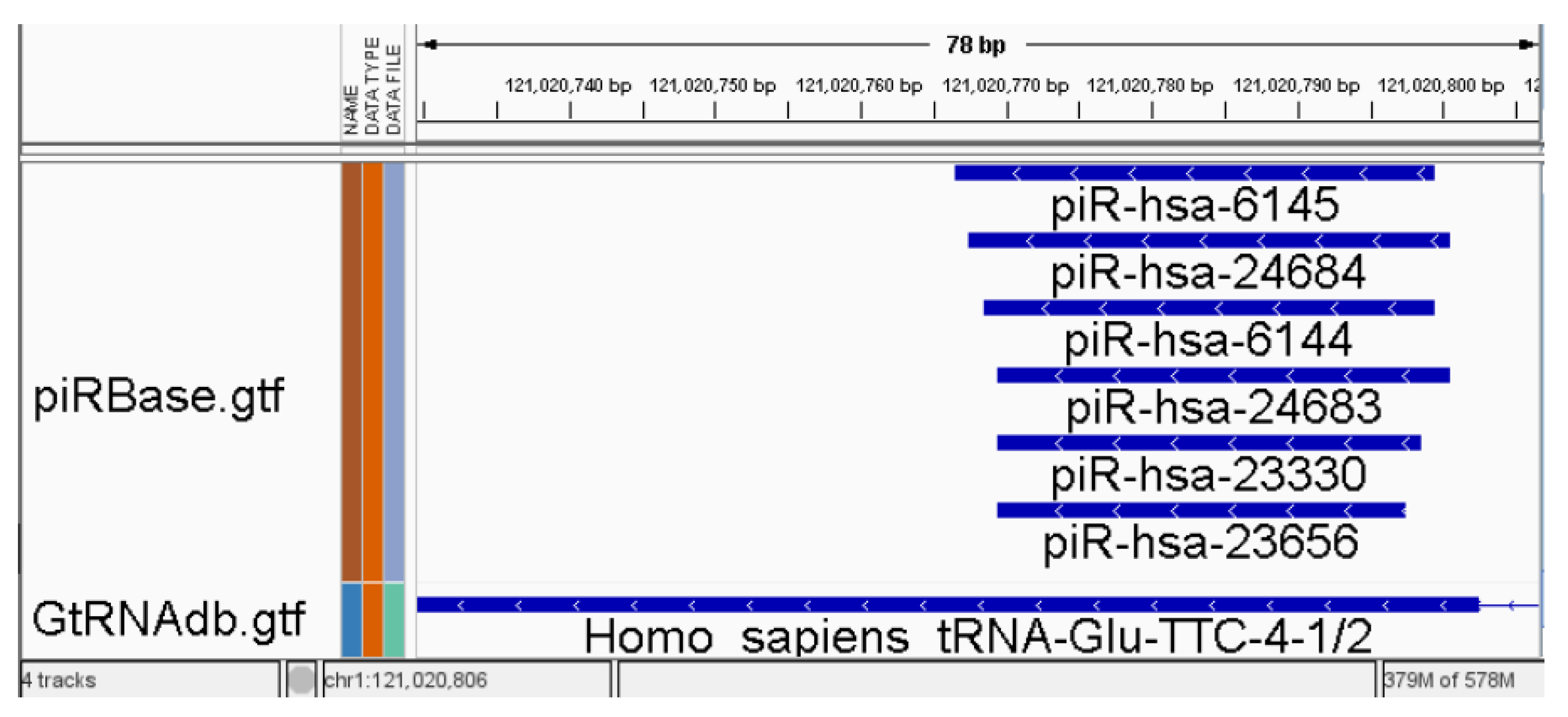
2.3.2. Merging of Overlapped Annotations Features
2.4. Using the Pipeline
2.4.1. Data Upload
2.4.2. Group Selection
2.4.3. Analysis Options
- Trimming—used adapters, size, and quality (QC) limits;
- BLAST—Switch taxa filter option for local database and number and size of the representative subsets. This selection is a tradeoff between resource consumption and the sensitivity of the pipeline to detect a rarely represented species in the sample. We recommend starting with a size of 200 reads, especially for a remote BLAST database and increasing if necessary.
- Differential expression—thresholds to filter expressible RNA (sequence in alignment-free analysis) and log2FoldChange and adjusted p-values to filter out statistically insignificant results.
- Strategy of the pipeline (Figure 2), where, in the case of “metagenome”, all reads at once will be mapped to generated on BLAST results metagenome or more traditional “successive” strategy, where, at first, samples mapped to the human genome and only reads unmapped to it will be mapped to the generated metagenome.
- Provide an email address and mail server if you prefer to receive notifications and report files on email.
2.4.4. Analysis Start
2.5. Filtering of Environmental Contamination
2.6. Source of Presented Small RNA Recognition
- Homo (9606);
- Bacteria (2);
- Fungi (4751);
- Viruses (10239);
- Archaea (2157);
- Amoebozoa (554915);
- Discoba (2611352);
- CRuMs (2608240);
- Metamonada (2611341);
- Sar (2698737);
- Eukaryota incertae sedis (2683617);
- Aphelida (2316435);
- Ichthyosporea (127916);
- Rotosphaerida (2686024);
- other sequences (28384).
2.7. Metagenome Generation and Alignment
2.8. Small RNA Types and Identified Species Catalogues
2.9. Differential Expression Analysis
2.10. Alignment-Free Sequence Analysis
2.11. Reports
- A consolidated Excel file report is presented, encompassing a comprehensive set of information (example report attached as Supplementary File S1):
- ○
- Analysis settings;
- ○
- Sample and trimming statistics;
- ○
- A catalogue of identified species;
- ○
- A catalogue of sRNA types;
- ○
- Counts of identified features;
- ○
- Spearman sample correlation tables with heatmap visualisation;
- ○
- Differential expression analysis for annotated RNA types;
- ○
- The file includes visualisations such as Volcano [50] and PCA plots.
- Quality Diagrams:
- Alignment-free Analysis (example report attached as Supplementary File S2):
- ○
- Results of the alignment-free analysis of RNA-seq data, featuring clustering and the initial identification of the RNA source, are presented in a separate Excel file.
- Post-translational Modifications and Enrichment Analysis, formatted in Excel for user convenience (example report attached as Supplementary File S3):
3. Results and Discussion
3.1. Merging of Overlapped Annotations Features
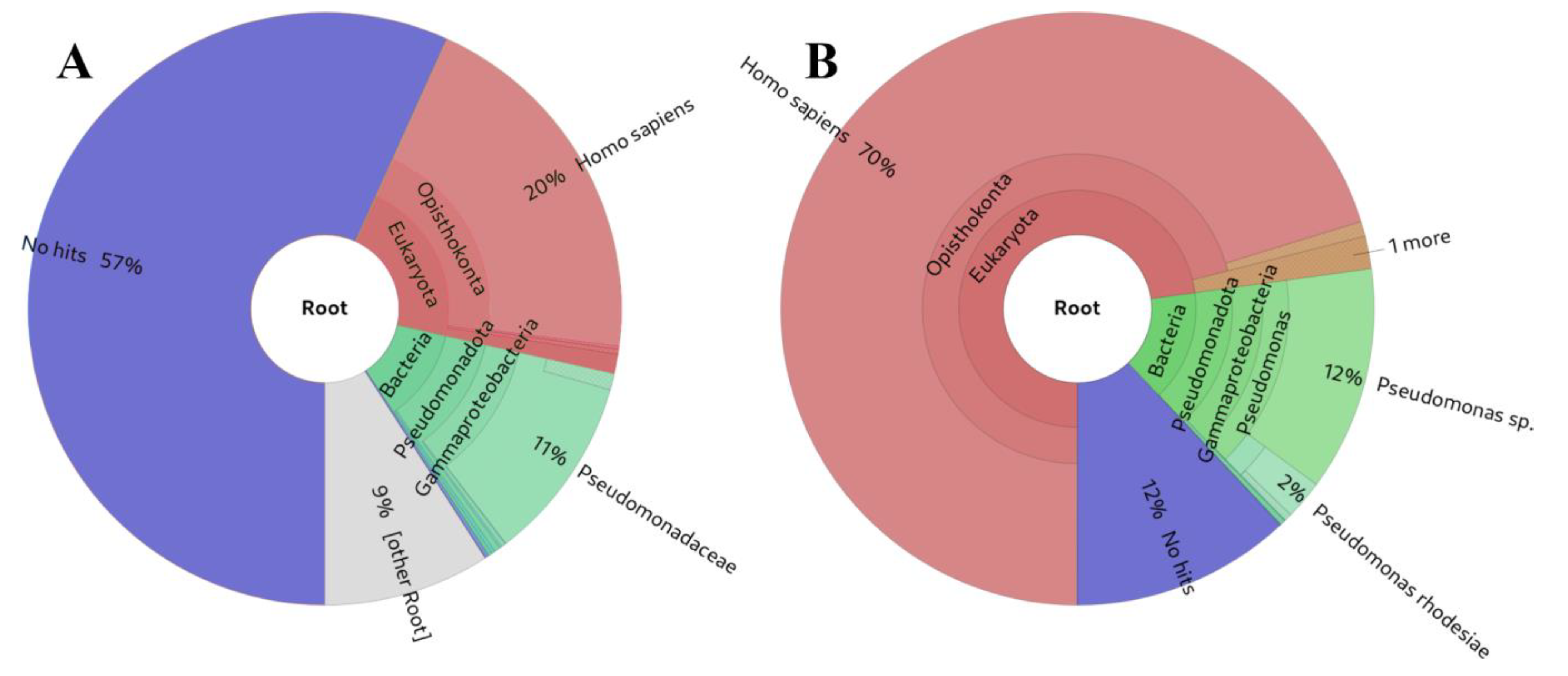
3.2. Testing the BLAST-Based Approach on Simulated Positive and Negative Controls
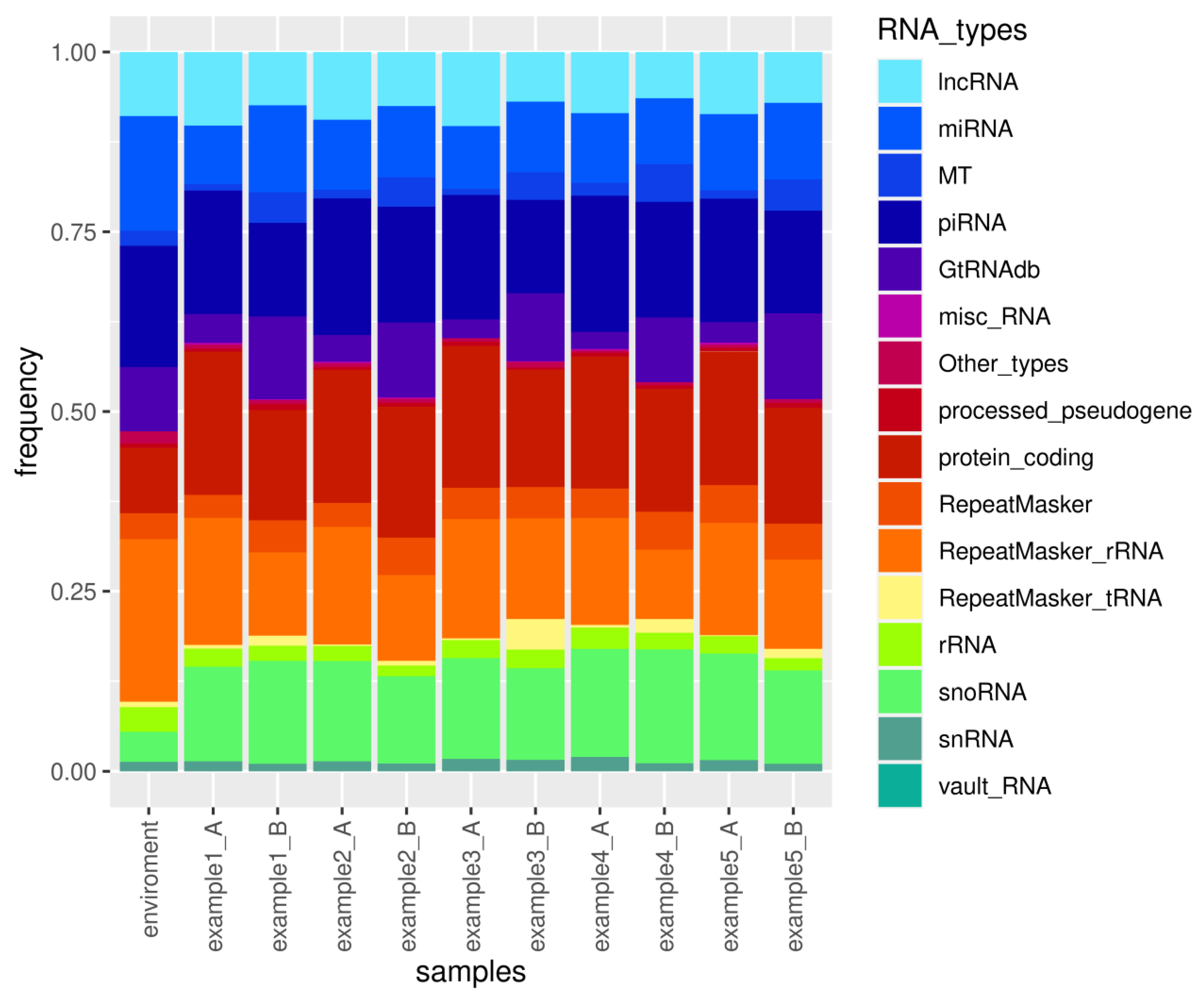
3.3. Example sRNAflow Reports on a Simulated Dataset
3.4. Comparison of Small RNA Analysis Pipelines on a Simulated Dataset
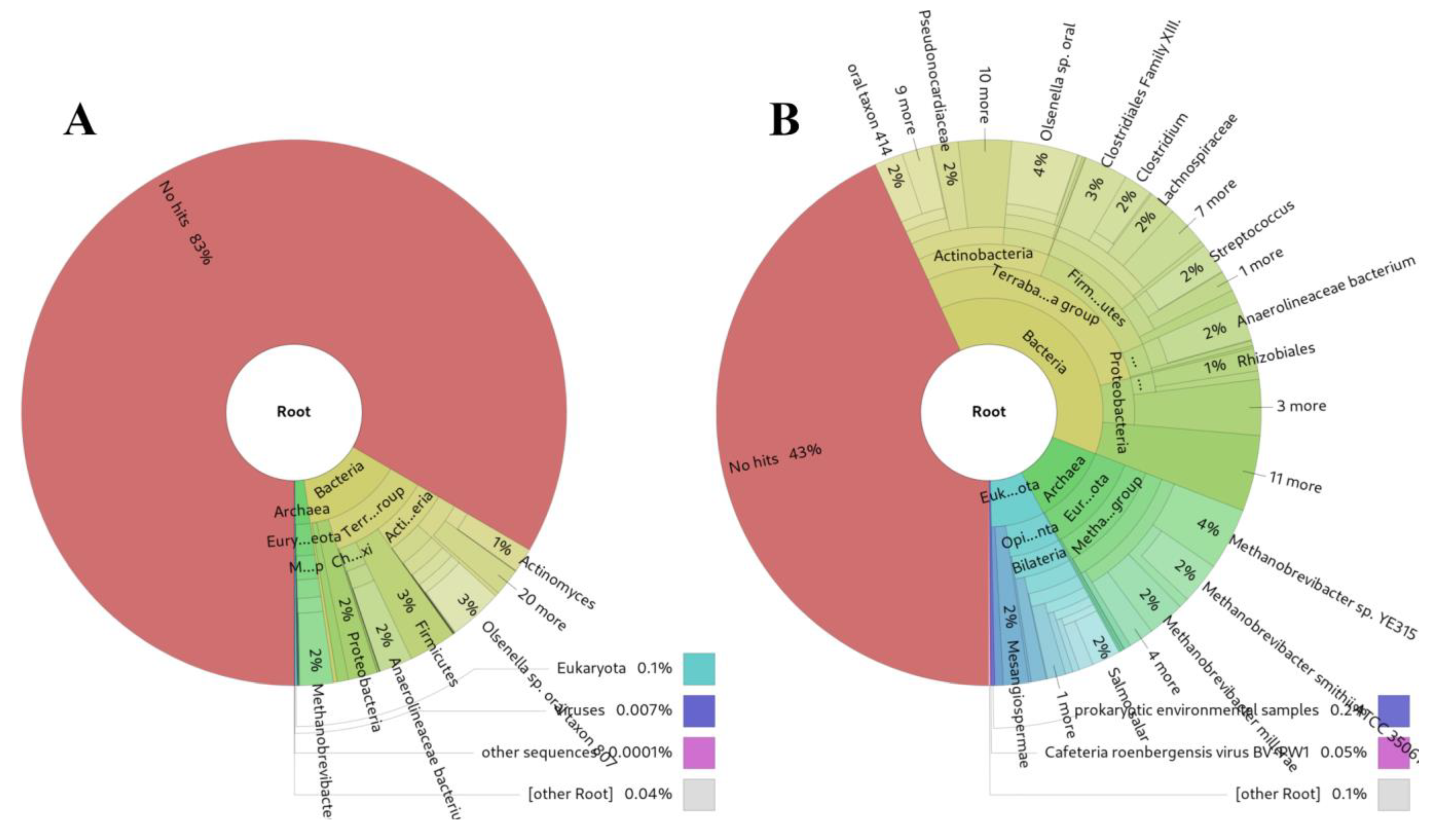
3.5. Analysis of Microbiome in Ancient DNA samples
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stefani, G.; Slack, F.J. Small Non-Coding RNAs in Animal Development. Nat. Rev. Mol. Cell Biol. 2008, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The Impact of microRNAs on Protein Output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Jaskiewicz, L.; Kolb, F.A.; Pillai, R.S. Post-Transcriptional Gene Silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 2005, 15, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA Pathway Provides an Adaptive Defense in the Transposon Arms Race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef]
- Christov, C.P.; Trivier, E.; Krude, T. Noncoding Human Y RNAs Are Overexpressed in Tumours and Required for Cell Proliferation. Br. J. Cancer 2008, 98, 981–988. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in Serum: A Novel Class of Biomarkers for Diagnosis of Cancer and Other Diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Boffelli, D.; Martin, D.I. Deep Sequencing of Serum Small RNAs Identifies Patterns of 5′ tRNA Half and YRNA Fragment Expression Associated with Breast Cancer. Biomark. Cancer 2014, 6, 37–47. [Google Scholar] [CrossRef]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Morici, L.; Splinter Bon-Durant, S.; Baddoo, M.; Lin, Z.; Fewell, C.; Taylor, C.M.; Flemington, E.K. Microbial Contamination in next Generation Sequencing: Implications for Sequence-Based Analysis of Clinical Samples. PLoS Pathog. 2014, 10, e1004437. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; McKenna, A.; Fennell, T.; Banks, E.; DePristo, M.; Getz, G. ContEst: Estimating Cross-Contamination of Human Samples in next-Generation Sequencing Data. Bioinformatics 2011, 27, 2601–2602. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Watson, M. Errors in RNA-Seq Quantification Affect Genes of Relevance to Human Disease. Genome Biol. 2015, 16, 177. [Google Scholar] [CrossRef]
- Zytnicki, M.; Gaspin, C. Mmannot: How to Improve Small-RNA Annotation? PLoS ONE 2020, 15, e0231738. [Google Scholar] [CrossRef] [PubMed]
- Barturen, G.; Rueda, A.; Hamberg, M.; Alganza, A.; Lebron, R.; Kotsyfakis, M.; Shi, B.-J.; Koppers-Lalic, D.; Hackenberg, M. sRNAbench: Profiling of Small RNAs and Its Sequence Variants in Single or Multi-Species High-Throughput Experiments. Meth. Next Gener. Seq. 2014, 1, 21–31. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Si, X.; Skogerbø, G.; Wang, J.; Cui, D.; Li, Y.; Sun, X.; Liu, L.; Sun, B.; Chen, R.; et al. piRBase: A Web Resource Assisting piRNA Functional Study. Database 2014, 2014, bau110. [Google Scholar] [CrossRef]
- Desvignes, T.; Loher, P.; Eilbeck, K.; Ma, J.; Urgese, G.; Fromm, B.; Sydes, J.; Aparicio-Puerta, E.; Barrera, V.; Espín, R.; et al. Unification of miRNA and isomiR Research: The mirGFF3 Format and the Mirtop API. Bioinformatics 2020, 36, 698–703. [Google Scholar] [CrossRef]
- The RNAcentral Consortium. RNAcentral: A Hub of Information for Non-Coding RNA Sequences. Nucleic Acids Res. 2019, 47, D221–D229. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb: A Database of Transfer RNA Genes Detected in Genomic Sequence. Nucleic Acids Res. 2009, 37, D93–D97. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Axtell, M.J. ShortStack: Comprehensive Annotation and Quantification of Small RNA Genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Sadovska, L.; Zayakin, P.; Bajo-Santos, C.; Endzeliņš, E.; Auders, J.; Keiša, L.; Jansons, J.; Lietuvietis, V.; Linē, A. Effects of Urinary Extracellular Vesicles from Prostate Cancer Patients on the Transcriptomes of Cancer-Associated and Normal Fibroblasts. BMC Cancer 2022, 22, 1055. [Google Scholar] [CrossRef]
- Jeske, T.; Huypens, P.; Stirm, L.; Höckele, S.; Wurmser, C.M.; Böhm, A.; Weigert, C.; Staiger, H.; Klein, C.; Beckers, J.; et al. DEUS: An R Package for Accurate Small RNA Profiling Based on Differential Expression of Unique Sequences. Bioinformatics 2019, 35, 4834–4836. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 Accurately Identifies Known and Hundreds of Novel microRNA Genes in Seven Animal Clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kim, T.-K.; Baxter, D.; Scherler, K.; Gordon, A.; Fong, O.; Etheridge, A.; Galas, D.J.; Wang, K. sRNAnalyzer-a Flexible and Customizable Small RNA Sequencing Data Analysis Pipeline. Nucleic Acids Res. 2017, 45, 12140–12151. [Google Scholar] [CrossRef]
- Pogorelcnik, R.; Vaury, C.; Pouchin, P.; Jensen, S.; Brasset, E. sRNAPipe: A Galaxy-Based Pipeline for Bioinformatic in-Depth Exploration of Small RNAseq Data. Mob. DNA 2018, 9, 25. [Google Scholar] [CrossRef]
- Rahman, R.-U.; Gautam, A.; Bethune, J.; Sattar, A.; Fiosins, M.; Magruder, D.S.; Capece, V.; Shomroni, O.; Bonn, S. Oasis 2: Improved Online Analysis of Small RNA-Seq Data. BMC Bioinform. 2018, 19, 54. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Gómez-Martín, C.; Giannoukakos, S.; Medina, J.M.; Scheepbouwer, C.; García-Moreno, A.; Carmona-Saez, P.; Fromm, B.; Pegtel, M.; Keller, A.; et al. sRNAbench and sRNAtoolbox 2022 Update: Accurate miRNA and sncRNA Profiling for Model and Non-Model Organisms. Nucleic Acids Res. 2022, 50, W710–W717. [Google Scholar] [CrossRef]
- Mjelle, R.; Aass, K.R.; Sjursen, W.; Hofsli, E.; Sætrom, P. sMETASeq: Combined Profiling of Microbiota and Host Small RNAs. iScience 2020, 23, 101131. [Google Scholar] [CrossRef]
- Kitchen, R. exceRpt: The Extra-Cellular RNA Processing Toolkit. Includes Software to Preprocess, Align, Quantitate, and Normalise smallRNA-Seq Datasets. Available online: https://rkitchen.github.io/exceRpt/ (accessed on 25 December 2023).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 25 December 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R Package Rsubread Is Easier, Faster, Cheaper and Better for Alignment and Quantification of RNA Sequencing Reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From Reads to Genes to Pathways: Differential Expression Analysis of RNA-Seq Experiments Using Rsubread and the edgeR Quasi-Likelihood Pipeline. F1000Res. 2016, 5, 1438. [Google Scholar]
- Urgese, G.; Paciello, G.; Acquaviva, A.; Ficarra, E. isomiR-SEA: An RNA-Seq Analysis Tool for miRNAs/isomiRs Expression Level Profiling and miRNA-mRNA Interaction Sites Evaluation. BMC Bioinform. 2016, 17, 148. [Google Scholar] [CrossRef]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. Msa: An R Package for Multiple Sequence Alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Easy Web Applications in R. Available online: http://www.rstudio.com/shiny (accessed on 29 December 2022).
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA Genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a Reference Set of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef]
- Piuco, R.; Galante, P.A.F. piRNAdb. Available online: https://www.pirnadb.org (accessed on 25 December 2023).
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 25 December 2023).
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-Line: Integrating Search and Context for Analysis of Transfer RNA Genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. Available online: https://bioconductor.org/packages/EnhancedVolcano (accessed on 25 December 2023).
- Zayakin, P.; Sadovska, L.; Eglītis, K.; Romanchikova, N.; Radoviča-Spalviņa, I.; Endzeliņš, E.; Liepniece-Karele, I.; Eglītis, J.; Linē, A. Extracellular Vesicles-A Source of RNA Biomarkers for the Detection of Breast Cancer in Liquid Biopsies. Cancers 2023, 15, 4329. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Whalen, V.M.; Goilav, B.; Max, K.E.A.; Tuschl, T. Cell and Microvesicle Urine microRNA Deep Sequencing Profiles from Healthy Individuals: Observations with Potential Impact on Biomarker Studies. PLoS ONE 2016, 11, e0147249. [Google Scholar] [CrossRef]
- Sadovska, L.; Zayakin, P.; Eglītis, K.; Endzeliņš, E.; Radoviča-Spalviņa, I.; Avotiņa, E.; Auders, J.; Keiša, L.; Liepniece-Karele, I.; Leja, M.; et al. Comprehensive Characterization of RNA Cargo of Extracellular Vesicles in Breast Cancer Patients Undergoing Neoadjuvant Chemotherapy. Front. Oncol. 2022, 12, 1005812. [Google Scholar] [CrossRef] [PubMed]
- Bajo-Santos, C.; Brokāne, A.; Zayakin, P.; Endzeliņš, E.; Soboļevska, K.; Belovs, A.; Jansons, J.; Sperga, M.; Llorente, A.; Radoviča-Spalviņa, I.; et al. Plasma and Urinary Extracellular Vesicles as a Source of RNA Biomarkers for Prostate Cancer in Liquid Biopsies. Front. Mol. Biosci. 2023, 10, 980433. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Zhou, H.; Zhang, P.; Song, T.; Ying, Z.; Yu, H.; Li, Y.; Zhao, Y.; Zeng, X.; et al. piRBase: Integrating piRNA Annotation in All Aspects. Nucleic Acids Res. 2022, 50, D265–D272. [Google Scholar] [CrossRef]
- Rosenkranz, D.; Zischler, H.; Gebert, D. piRNAclusterDB 2.0: Update and Expansion of the piRNA Cluster Database. Nucleic Acids Res. 2022, 50, D259–D264. [Google Scholar] [CrossRef]
- Tosar, J.P.; Rovira, C.; Cayota, A. Non-Coding RNA Fragments Account for the Majority of Annotated piRNAs Expressed in Somatic Non-Gonadal Tissues. Commun. Biol. 2018, 1, 2. [Google Scholar] [CrossRef]
- Frazee, A.C.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Polyester: Simulating RNA-Seq Datasets with Differential Transcript Expression. Bioinformatics 2015, 31, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Grimm, D.G. Design and Analysis of RNA Sequencing Data. In Next Generation Sequencing and Data Analysis; Springer International Publishing: Cham, Switzerland, 2021; pp. 143–175. ISBN 9783030624897. [Google Scholar]
- Costa-Silva, J.; Domingues, D.S.; Menotti, D.; Hungria, M.; Lopes, F.M. Temporal Progress of Gene Expression Analysis with RNA-Seq Data: A Review on the Relationship between Computational Methods. Comput. Struct. Biotechnol. J. 2023, 21, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic Microbial Community Profiling Using Unique Clade-Specific Marker Genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef]
- Kazarina, A.; Petersone-Gordina, E.; Kimsis, J.; Kuzmicka, J.; Zayakin, P.; Griškjans, Ž.; Gerhards, G.; Ranka, R. The Postmedieval Latvian Oral Microbiome in the Context of Modern Dental Calculus and Modern Dental Plaque Microbial Profiles. Genes 2021, 12, 309. [Google Scholar] [CrossRef]
- Ķimsis, J.; Pokšāne, A.; Kazarina, A.; Vilcāne, A.; Petersone-Gordina, E.; Zayakin, P.; Gerhards, G.; Ranka, R. Tracing Microbial Communities Associated with Archaeological Human Samples in Latvia, 7–11th Centuries AD. Environ. Microbiol. Rep. 2023, 15, 383–391. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Version | Accessed | Format | Features | Merged | % Merged |
|---|---|---|---|---|---|---|
| miRBase_hairpin | v22 | December 2013 | GFF3 | 1918 | 1859 | 3 |
| miRBase_mature | December 2013 | GFF3 | 2883 | 2813 | 2 | |
| GtRNAdb | v21 | December 2013 | FASTA | 432 | 432 | 0 |
| LNCipedia | v5.2 | December 2013 | GTF | 357,620 | 151,562 | 58 |
| LNCipedia_hc | December 2013 | GTF | 288,174 | 127,290 | 56 | |
| piRNAdb | v1.7.6 | December 2013 | FASTA | 814,994 | 558,329 | 31 |
| piRBase | v1 | December 2013 | FASTA | 797,231 | 549,328 | 31 |
| Ensembl | GRCh38.p14 | December 2013 | GTF | 1,649,690 | 345,110 | 79 |
| miRNA | 1879 | 1822 | 3 | |||
| rRNA | 53 | 53 | 0 | |||
| protein_coding | 1,387,673 | 235,196 | 83 | |||
| processed_pseudogene | 11,773 | 11,731 | 0 | |||
| snRNA | 1910 | 1910 | 0 | |||
| snoRNA | 942 | 925 | 2 | |||
| MT | 37 | 32 | 14 | |||
| lncRNA | 217,724 | 71,419 | 67 | |||
| vault_RNA | 1 | 1 | 0 | |||
| YRNA | 814 | 814 | 0 | |||
| notY_misc_RNA | 1407 | 1407 | 0 | |||
| Other_types | 25,477 | 19,800 | 22 | |||
| RepeatMasker | Gencode v44 | December 2013 | FASTA | 5,683,690 | 5,536,563 | 3 |
| RepeatMasker_tRNA | 2164 | 2164 | 0 | |||
| RepeatMasker_rRNA | 565 | 538 | 5 |
| Pipeline | Filtered QC and <15 bp | Filtered Environment | Annotated Human | Ambiguous Human | Unannotated Human | Identified Other Species | Unidentified |
|---|---|---|---|---|---|---|---|
| sMETASeq (RNACentral) | 14% | - | 1% | 17% | 41% | 5% | 22% |
| sMETASeq (MiRBase) | 14% | - | 6% | 0.01% | 52% | 5% | 22% |
| Cutadapt + Kraken2 | 14% | - | - | - | 13% | 13% | 59% |
| Cutadapt + bowtie2 + Rsubread(Ens.) + Kraken2 | 14% | - | 15% | 12% | 25% | 6% | 26% |
| exceRpt | 36% | - | 30% | 4% | - | 30% | |
| sRNAtoolbox | 14% | - | 42% | 14% | 17% | 9 * + 4% | |
| sRNAflow (metagenome) | 14% | - | 44% | 0% | 5% | 16% | 19% |
| sRNAflow (successively) | 14% | - | 46% | 0% | 6% | 14% | 19% |
| Pipelines that include filtering against an environmental sample | |||||||
| Cutadapt + Kraken2 | 14% | 28% | - | - | 7% | 11% | 40% |
| Cutadapt + bowtie2 + Rsubread(Ens.) + Kraken2 | 14% | 28% | 7% | 5% | 15% | 6% | 25% |
| sRNAflow (metagenome) | 14% | 28% | 19% | 0% | 6% | 15% | 18% |
| sRNAflow (successively) | 14% | 28% | 21% | 0% | 6% | 13% | 18% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayakin, P. sRNAflow: A Tool for the Analysis of Small RNA-Seq Data. Non-Coding RNA 2024, 10, 6. https://doi.org/10.3390/ncrna10010006
Zayakin P. sRNAflow: A Tool for the Analysis of Small RNA-Seq Data. Non-Coding RNA. 2024; 10(1):6. https://doi.org/10.3390/ncrna10010006
Chicago/Turabian StyleZayakin, Pawel. 2024. "sRNAflow: A Tool for the Analysis of Small RNA-Seq Data" Non-Coding RNA 10, no. 1: 6. https://doi.org/10.3390/ncrna10010006
APA StyleZayakin, P. (2024). sRNAflow: A Tool for the Analysis of Small RNA-Seq Data. Non-Coding RNA, 10(1), 6. https://doi.org/10.3390/ncrna10010006