
RNA N6-Methyladenosine Affects Copper-Induced Oxidative Stress Response in Arabidopsis thaliana

 , and
, and 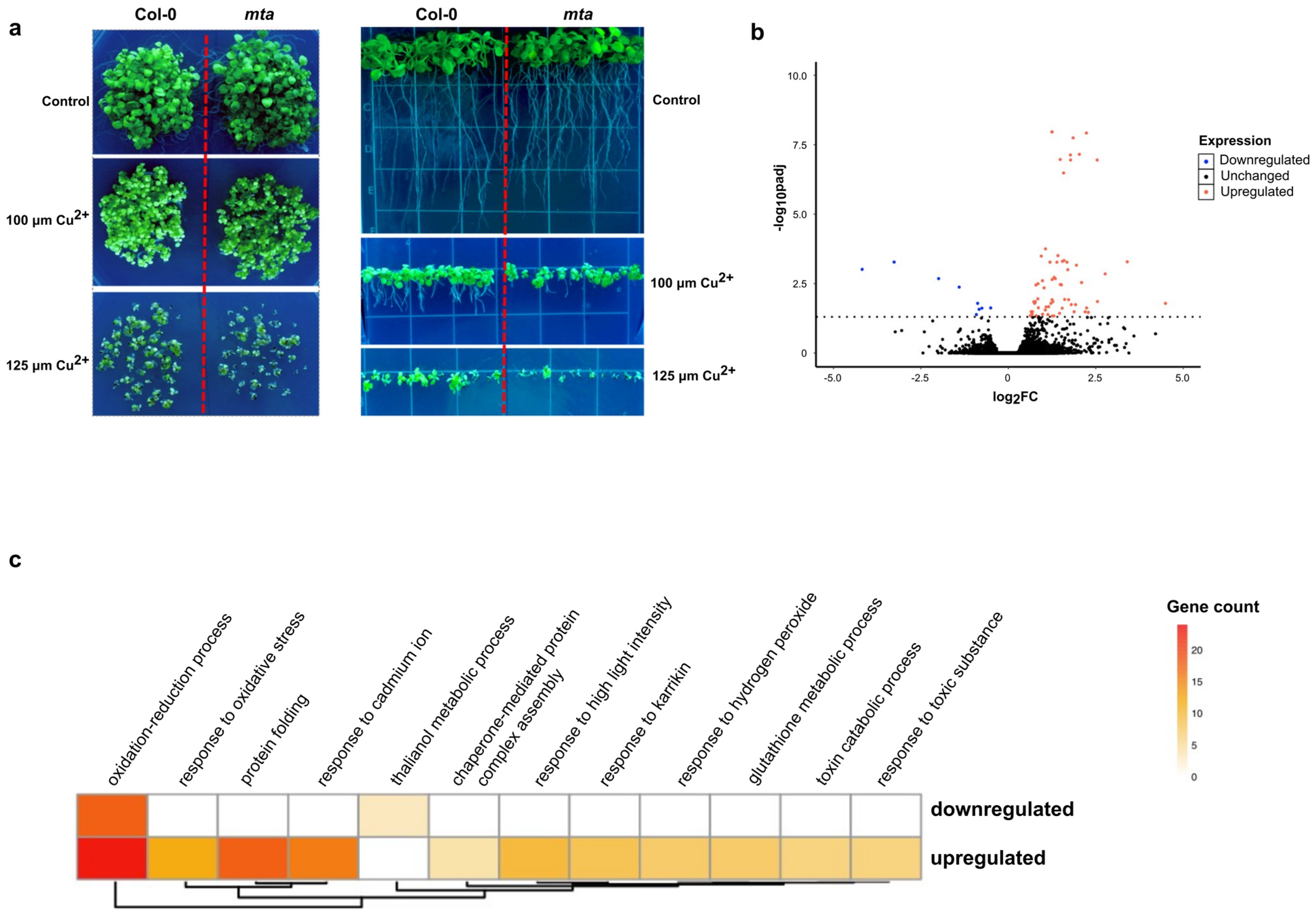{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Reduction in Global m6A Levels Due to Loss of MTA Leads to Lack of Response to Copper-Induced Oxidative Stress
2.2. Copper-Induced Stress Increases the Abundance of Transcripts Encoding Proteins Involved in Responses to Oxidative Stress and Pathogen Defense
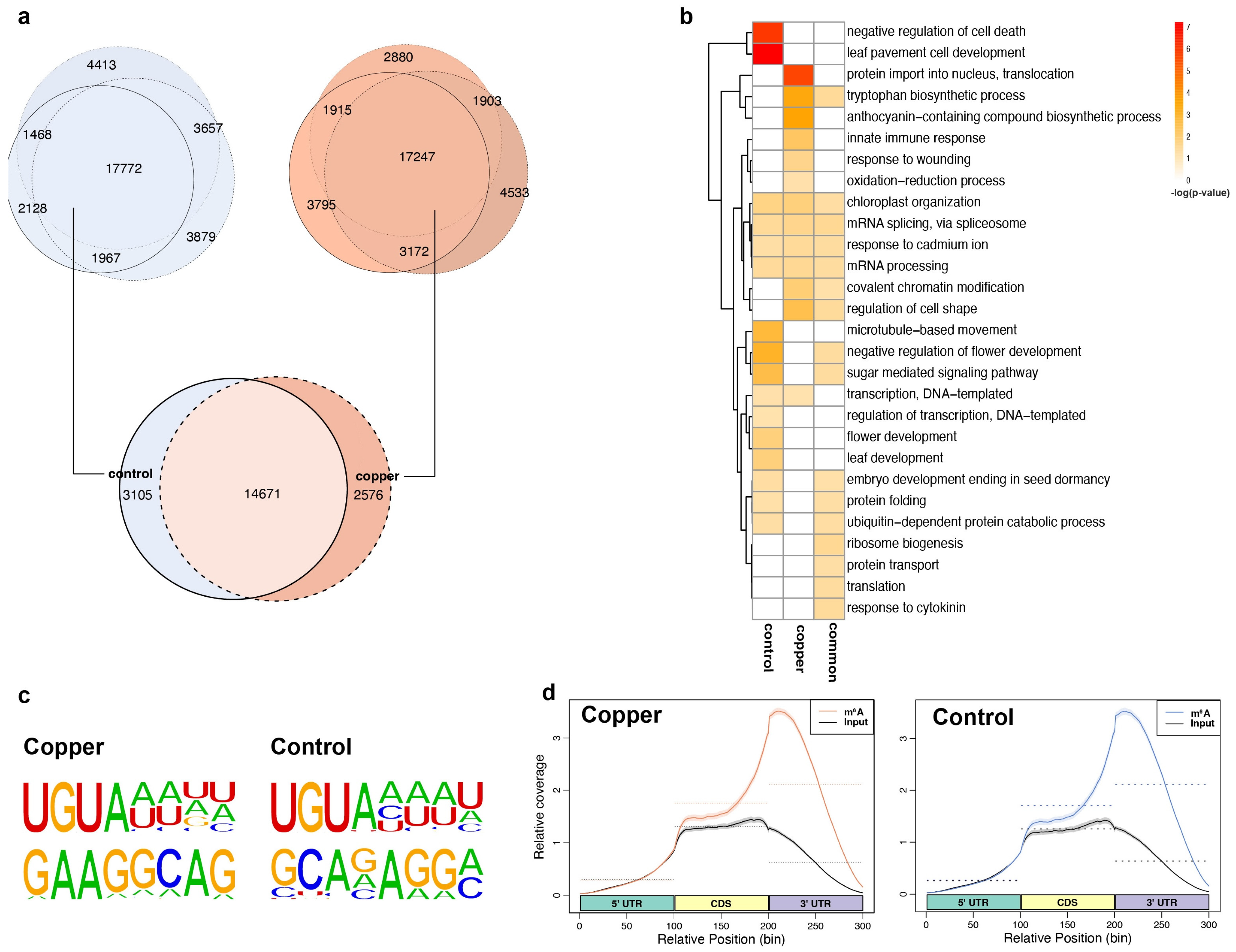
2.3. Thousands of m6A Peaks Are Specifically Induced or Depleted upon Copper-Induced Oxidative Stress in mRNA
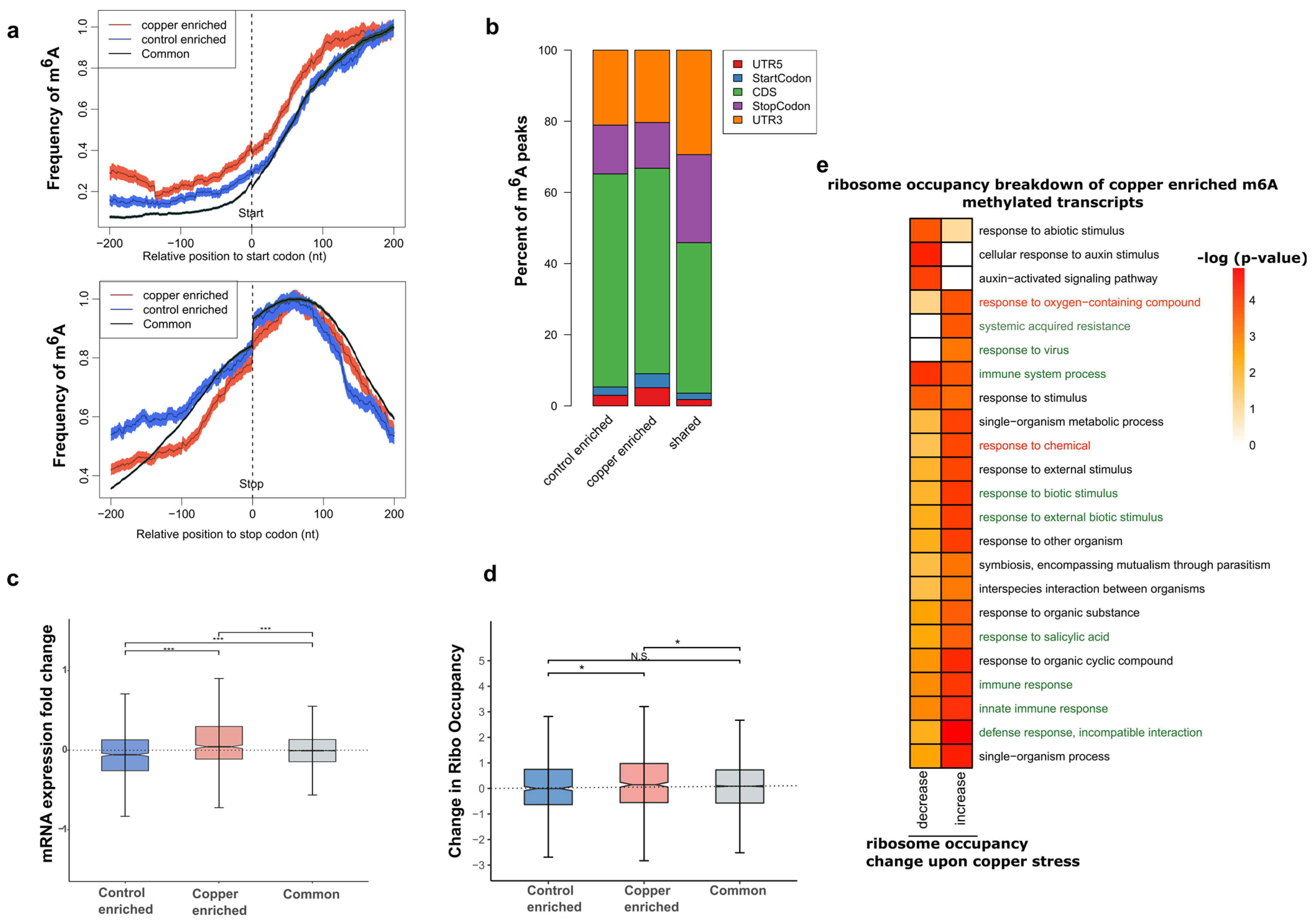
2.4. m6A Tends to Be Localized in mRNA 3′ UTRs and Its Localization Pattern Is Sensitive to Copper-Induced Oxidative Stress
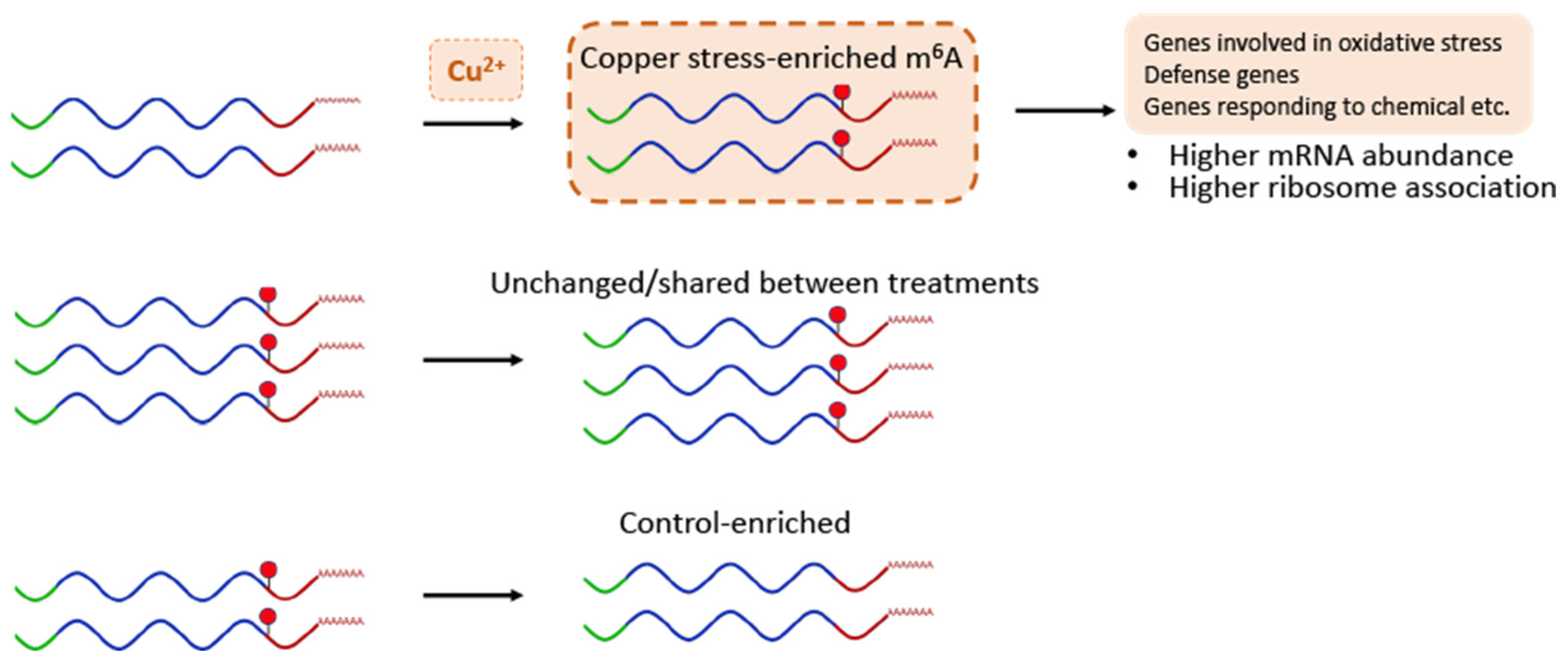
2.5. Transcripts That Contain Copper Stress-Enriched m6A Are More Likely to Show Increased mRNA Levels
2.6. Transcripts with Copper Stress-Induced m6A Are More Likely to Be Polysome Associated
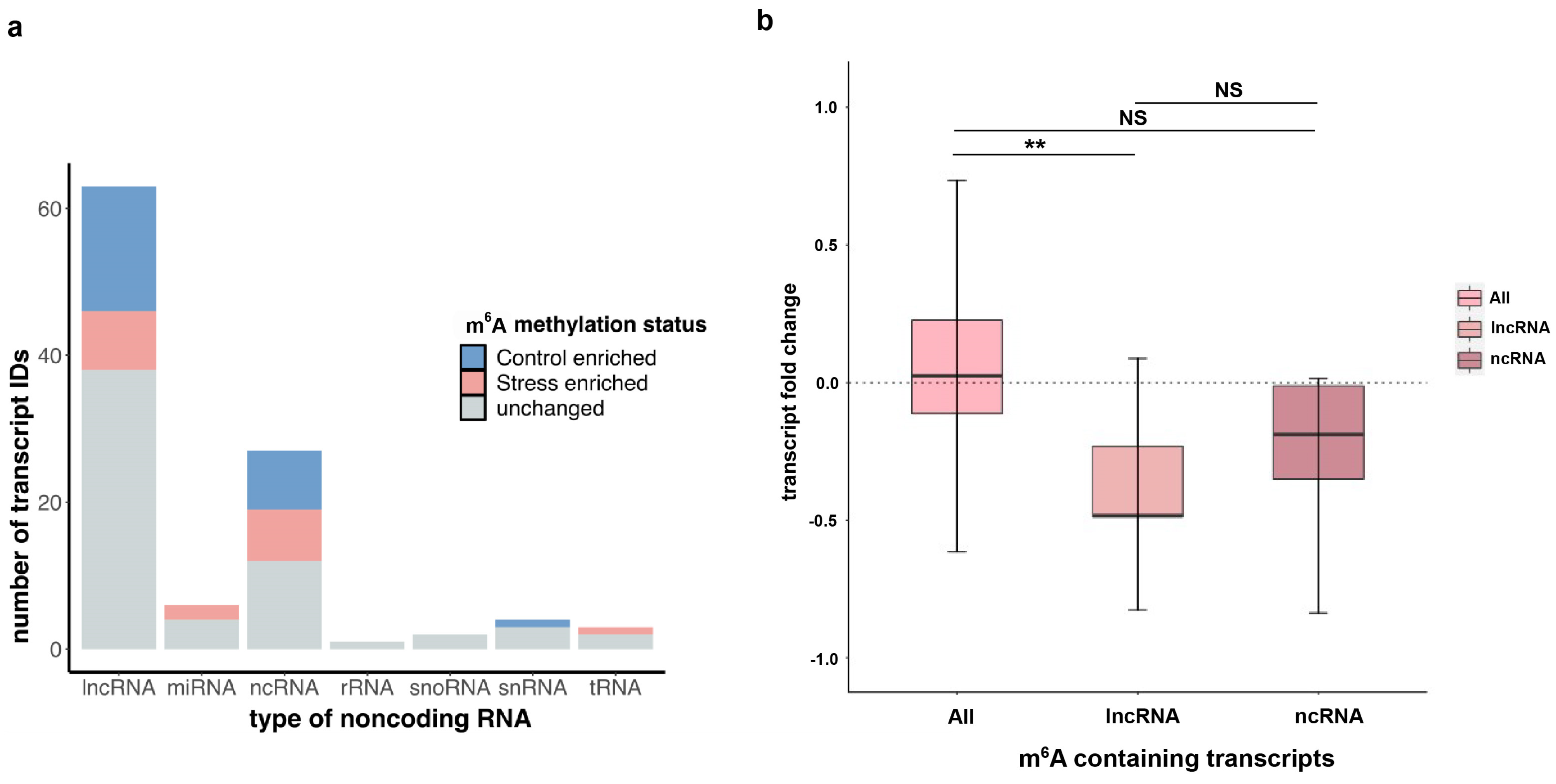
2.7. Long Non-Coding RNAs (lncRNAs) and m6A Association
3. Discussion
4. Materials and Methods
4.1. Stress Treatment for RNA-Seq, m6A-IP-Seq, and Polysomal RNA-Seq
4.2. m6A RNA Immunoprecipitation and Sequencing (m6A-IP-Seq)
4.3. Polysomal RNA-Sequencing
4.4. Bioinformatics Analysis of RNA-Seq, m6A-IP-Seq, and Polysome-Seq Data
4.4.1. Read Processing
4.4.2. Differential RNA Abundance
4.4.3. m6A Peak Calling
4.4.4. Motif Searches
4.4.5. m6A Frequency
4.4.6. Polysome Occupancy
4.4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartels, D.; Sunkar, R. Drought and Salt Tolerance in Plants. Crit. Rev. Plant Sci. 2005, 24, 23–58. [Google Scholar] [CrossRef]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Munns, R.; Millar, A.H. Seven plant capacities to adapt to abiotic stress. J. Exp. Bot. 2023, 74, 4308–4323. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Zandalinas, S.I.; Fichman, Y.; Van Breusegem, F. Reactive oxygen species signalling in plant stress responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Seo, P.J.; Lee, H.-J.; Park, C.-M. A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 2012, 70, 831–844. [Google Scholar] [CrossRef]
- Yang, C.; Li, W.; Cao, J.; Meng, F.; Yu, Y.; Huang, J.; Jiang, L.; Liu, M.; Zhang, Z.; Chen, X.; et al. Activation of ethylene signaling pathways enhances disease resistance by regulating ROS and phytoalexin production in rice. Plant J. 2017, 89, 338–353. [Google Scholar] [CrossRef]
- Kawarazaki, T.; Kimura, S.; Iizuka, A.; Hanamata, S.; Nibori, H.; Michikawa, M.; Imai, A.; Abe, M.; Kaya, H.; Kuchitsu, K. A low temperature-inducible protein AtSRC2 enhances the ROS-producing activity of NADPH oxidase AtRbohF. Biochim. Biophys. Acta BBA Mol. Cell Res. 2013, 1833, 2775–2780. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.-K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Jagadeeswaran, G.; Li, Y.-F.; Sunkar, R. Redox signaling mediates the expression of a sulfate-deprivation-inducible microRNA395 in Arabidopsis. Plant J. 2014, 77, 85–96. [Google Scholar] [CrossRef]
- Zhou, H.; Finkemeier, I.; Guan, W.; Tossounian, M.-A.; Wei, B.; Young, D.; Huang, J.; Messens, J.; Yang, X.; Zhu, J.; et al. Oxidative stress-triggered interactions between the succinyl- and acetyl-proteomes of rice leaves. Plant Cell Environ. 2018, 41, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhang, H.; Yu, B.; Xiong, L.; Xia, Y. Proteomic identification of early salicylate- and flg22-responsive redox-sensitive proteins in Arabidopsis. Sci. Rep. 2015, 5, 8625. [Google Scholar] [CrossRef]
- Jing, M.; Zhang, H.; Wei, M.; Tang, Y.; Xia, Y.; Chen, Y.; Shen, Z.; Chen, C. Reactive Oxygen Species Partly Mediate DNA Methylation in Responses to Different Heavy Metals in Pokeweed. Front. Plant Sci. 2022, 13, 845108. [Google Scholar] [CrossRef]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef]
- Cheng, P.; Bao, S.; Li, C.; Tong, J.; Shen, L.; Yu, H. RNA N6-methyladenosine modification promotes auxin biosynthesis required for male meiosis in rice. Dev. Cell 2022, 57, 246–259.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.C.; Janssen, K.A.; Palos, K.; Nelson, A.D.L.; Vandivier, L.E.; Garcia, B.A.; Lyons, E.; Beilstein, M.A.; Gregory, B.D. N6-methyladenosine and RNA secondary structure affect transcript stability and protein abundance during systemic salt stress in Arabidopsis. Plant Direct 2020, 4, e00239. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef]
- Flamand, M.N.; Meyer, K.D. m6A and YTHDF proteins contribute to the localization of select neuronal mRNAs. Nucleic Acids Res. 2022, 50, 4464–4483. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Sun, J.; Wu, B.; Gao, Y.; Nie, H.; Nie, Z.; Quan, S.; Wang, Y.; Cao, X.; Li, S. CPSF30-L-mediated recognition of mRNA m6A modification controls alternative polyadenylation of nitrate signaling-related gene transcripts in Arabidopsis. Mol. Plant 2021, 14, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, Y.-S.; Ping, X.-L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.-Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Govindan, G.; Sharma, B.; Li, Y.; Armstrong, C.D.; Merum, P.; Rohila, J.S.; Gregory, B.D.; Sunkar, R. mRNA N6-methyladenosine is critical for cold tolerance in Arabidopsis. Plant J. 2022, 111, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.-Y.; Xu, T.; Kang, H. N6-Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 39, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.S.; Bielewicz, D.; Gulanicz, T.; Bodi, Z.; Yu, X.; Anderson, S.J.; Szewc, L.; Bajczyk, M.; Dolata, J.; Grzelak, N.; et al. mRNA adenosine methylase (MTA) deposits m6A on pri-miRNAs to modulate miRNA biogenesis in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2020, 117, 21785–21795. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef]
- Sunkar, R.; Bartels, D.; Kirch, H.-H. Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J. 2003, 35, 452–464. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, B.; Wu, T.; Ding, Y.; Ding, X.; Chu, Z. Copper Ion Elicits Defense Response in Arabidopsis thaliana by Activating Salicylate- and Ethylene-Dependent Signaling Pathways. Mol. Plant 2015, 8, 1550–1553. [Google Scholar] [CrossRef]
- Drazkiewicz, M.; Skórzyńska-Polit, E.; Krupa, Z. Copper-induced oxidative stress and antioxidant defence in Arabidopsis thaliana. Biometals 2004, 17, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Bourdais, G.; Burdiak, P.; Gauthier, A.; Nitsch, L.; Salojärvi, J.; Rayapuram, C.; Idänheimo, N.; Hunter, K.; Kimura, S.; Merilo, E.; et al. Large-Scale Phenomics Identifies Primary and Fine-Tuning Roles for CRKs in Responses Related to Oxidative Stress. PLoS Genet. 2015, 11, e1005373. [Google Scholar] [CrossRef]
- Sappl, P.G.; Carroll, A.J.; Clifton, R.; Lister, R.; Whelan, J.; Harvey Millar, A.; Singh, K.B. The Arabidopsis glutathione transferase gene family displays complex stress regulation and co-silencing multiple genes results in altered metabolic sensitivity to oxidative stress. Plant J. 2009, 58, 53–68. [Google Scholar] [CrossRef]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.D.L.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-Methyladenosine Inhibits Local Ribonucleolytic Cleavage to Stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157.e3. [Google Scholar] [CrossRef]
- Wei, L.-H.; Song, P.; Wang, Y.; Lu, Z.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.-C.; Jia, G. The m6A Reader ECT2 Controls Trichome Morphology by Affecting mRNA Stability in Arabidopsis. Plant Cell 2018, 30, 968–985. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, J.; Lian, B.; Gu, H.; Li, Y.; Qi, Y. Global identification of Arabidopsis lncRNAs reveals the regulation of MAF4 by a natural antisense RNA. Nat. Commun. 2018, 9, 5056. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Zheng, Y.; Vemireddy, L.R.; Panda, S.K.; Jose, S.; Ranjan, A.; Panda, P.; Govindan, G.; Cui, J.; Wei, K.; et al. Comparative transcriptome and translatome analysis in contrasting rice genotypes reveals differential mRNA translation in salt-tolerant Pokkali under salt stress. BMC Genom. 2018, 19, 935. [Google Scholar] [CrossRef]
- D’Antonio, M.; D’Onorio De Meo, P.; Pallocca, M.; Picardi, E.; D’Erchia, A.M.; Calogero, R.A.; Castrignanò, T.; Pesole, G. RAP: RNA-Seq Analysis Pipeline, a new cloud-based NGS web application. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, B.; Govindan, G.; Li, Y.; Sunkar, R.; Gregory, B.D. RNA N6-Methyladenosine Affects Copper-Induced Oxidative Stress Response in Arabidopsis thaliana. Non-Coding RNA 2024, 10, 8. https://doi.org/10.3390/ncrna10010008
Sharma B, Govindan G, Li Y, Sunkar R, Gregory BD. RNA N6-Methyladenosine Affects Copper-Induced Oxidative Stress Response in Arabidopsis thaliana. Non-Coding RNA. 2024; 10(1):8. https://doi.org/10.3390/ncrna10010008
Chicago/Turabian StyleSharma, Bishwas, Ganesan Govindan, Yongfang Li, Ramanjulu Sunkar, and Brian D. Gregory. 2024. "RNA N6-Methyladenosine Affects Copper-Induced Oxidative Stress Response in Arabidopsis thaliana" Non-Coding RNA 10, no. 1: 8. https://doi.org/10.3390/ncrna10010008
APA StyleSharma, B., Govindan, G., Li, Y., Sunkar, R., & Gregory, B. D. (2024). RNA N6-Methyladenosine Affects Copper-Induced Oxidative Stress Response in Arabidopsis thaliana. Non-Coding RNA, 10(1), 8. https://doi.org/10.3390/ncrna10010008