Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops


Abstract
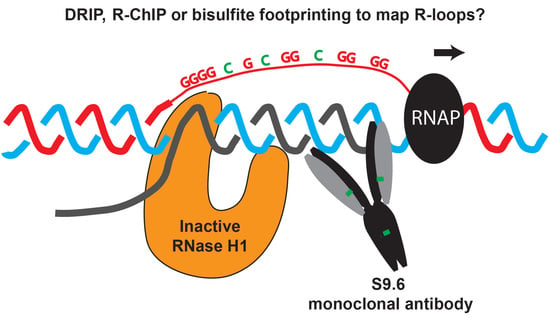
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Non-Denaturing Bisulfite Footprinting to Map R-Loops
3. The S9.6-Based Methods to Map and Quantify R-Loops
3.1. Quantifying R-Loop Formation Using Dot Blots
3.2. Visualizing R-Loop Formation Using Immunofluorescence Approaches
3.3. DNA:RNA Immunoprecipitation (DRIP) and DRIP-Like Methods
3.3.1. Apparent Lack of Robustness of the DRIP Method
3.3.2. Factors Contributing to the Lack of Robustness of DRIP
4. RNase H1-Based Methods to Map R-Loops
5. RNase H Over-Expression as a Tool to Probe R-Loop Functions
6. Conclusions
- Current immunofluorescence approaches are unlikely to be a reliable way of quantifying R-loops.
- The affinity of the S9.6 antibody for RNA:RNA hybrids and other non-B forms of DNA is potentially a confounding parameter for the quantification and the mapping of genuine R-loops using DRIP-like approaches.
- The possible influence of the cell-cycle on the formation and/or stability of R-loops and the fact that the S9.6 antibody could theoretically recognize DNA:RNA hybrids associated with Okazaki fragments suggest that the cell-cycle profile of the starting material is a parameter to consider when performing DRIP.
- More evidence is needed to demonstrate that R-ChIP is able to map all types of R-loops rather than a subset of abundant, G-skewed, promoter-associated R-loops.
- Possible indirect perturbations to the transcriptome and proteome associated with prolonged manipulation of RNase H levels must be taken into consideration when using this strategy to characterize R-loop functions.
Acknowledgments
Conflicts of Interest
References
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chedin, F. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Manley, J.L. R loops and links to human disease. J. Mol. Biol. 2016, 429, 3168–3180. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Mol. Cell 2017, 68, 745–757.e5. [Google Scholar] [CrossRef] [PubMed]
- Dumelie, J.G.; Jaffrey, S.R. Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. eLife 2017, 6, e28306. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Xu, H.; Li, K.; Fan, Y.; Liu, Y.; Yang, X.; Sun, Q. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat. Plants 2017, 3, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chiang, H.C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 regulates transcription elongation by RNA polymerase ii to prevent R-loop accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.; Zofall, M.; Balachandran, V.; Thillainadesan, G.; Sugiyama, T.; Wheeler, D.; Zhou, M.; Grewal, S.I. SNF2 family protein FFT3 suppresses nucleosome turnover to promote epigenetic inheritance and proper replication. Mol. Cell 2017, 66, 50–62.e6. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Moyano, E.; Mergui, X.; Garcia-Rubio, M.L.; Barroso, S.; Aguilera, A. The yeast and human fact chromatin-reorganizing complexes solve R-loop-mediated transcription-replication conflicts. Genes Dev. 2014, 28, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pichardo, D.; Canas, J.C.; Garcia-Rubio, M.L.; Gomez-Gonzalez, B.; Rondon, A.G.; Aguilera, A. Histone mutants separate R loop formation from genome instability induction. Mol. Cell 2017, 66, 597–609.e595. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, J.R.; Zhang, J. Nascent RNA folding mitigates transcription-associated mutagenesis. Genome Res. 2016, 26, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, A.; Grosso, A.R.; Elkaoutari, A.; Coleno, E.; Presle, A.; Sridhara, S.C.; Janbon, G.; Geli, V.; de Almeida, S.F.; Palancade, B. Introns protect eukaryotic genomes from transcription-associated genetic instability. Mol. Cell 2017, 67, 608–621.e606. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of RNA–DNA hybrids and associated chromosome instability. eLife 2013, 2, e00505. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.e719. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Hall, A.N.; Merrikh, C.N.; Ragheb, M.; Tabakh, H.; Pollock, A.J.; Woodward, J.J.; Dreifus, J.E.; Merrikh, H. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell 2017, 170, 787–799.e718. [Google Scholar] [CrossRef] [PubMed]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Z.; Pannunzio, N.R.; Han, L.; Hsieh, C.L.; Yu, K.; Lieber, M.R. The strength of an ig switch region is determined by its ability to drive R loop formation and its number of WGCW sites. Cell Rep. 2014, 8, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Clayton, D.A. Initiation of mitochondrial DNA replication by transcription and R-loop processing. J. Biol. Chem. 1998, 273, 30614–30621. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol. Cell 2018, 69, 426–437.e427. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Puget, N.; Lin, Y.L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Giri, P.K.; Kazadi, D.; Laffleur, B.; Zhang, W.; Grinstein, V.; Pefanis, E.; Brown, L.M.; Ladewig, E.; Martin, O.; et al. Nuclear proximity of Mtr4 to RNA exosome restricts DNA mutational asymmetry. Cell 2017, 169, 523–537.e515. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5, e17548. [Google Scholar] [CrossRef] [PubMed]
- Boguslawski, S.J.; Smith, D.E.; Michalak, M.A.; Mickelson, K.E.; Yehle, C.O.; Patterson, W.L.; Carrico, R.J. Characterization of monoclonal antibody to DNA·RNA and its application to immunodetection of hybrids. J. Immunol. Methods 1986, 89, 123–130. [Google Scholar] [CrossRef]
- Konig, F.; Schubert, T.; Langst, G. The monoclonal S9.6 antibody exhibits highly variable binding affinities towards different R-loop sequences. PLoS ONE 2017, 12, e0178875. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.D.; Garboczi, D.N.; Singh, K.; Hu, Z.; Leppla, S.H.; Leysath, C.E. The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J. Mol. Recognit. 2013, 26, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Hartono, S.R.; Malapert, A.; Legros, P.; Bernard, P.; Chedin, F.; Vanoosthuyse, V. The affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-loops in fission yeast. J. Mol. Biol. 2018, 430, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.S.; Viscidi, R.P.; Vonderfecht, S.L.; Eiden, J.J.; Yolken, R.H. Monoclonal antibody assay for detection of double-stranded RNA and application for detection of group a and non-group a rotaviruses. J. Clin. Microbiol. 1989, 27, 6–12. [Google Scholar] [PubMed]
- Halasz, L.; Karanyi, Z.; Boros-Olah, B.; Kuik-Rozsa, T.; Sipos, E.; Nagy, E.; Mosolygo, L.A.; Mazlo, A.; Rajnavolgyi, E.; Halmos, G.; et al. RNA–DNA hybrid (R-loop) immunoprecipitation mapping: An analytical workflow to evaluate inherent biases. Genome Res. 2017, 27, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Hou, Q.; Cheng, L.; Xu, W.; Hong, Y.; Li, S.; Sun, Q. RNase H1 cooperates with DNA gyrases to restrict R-loops and maintain genome integrity in arabidopsis chloroplasts. Plant Cell 2017, 29, 2478–2497. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Sun, H.; De Hoyos, C.L.; Bailey, J.K.; Liang, X.H.; Crooke, S.T. Dynamic nucleoplasmic and nucleolar localization of mammalian RNase H1 in response to RNap I transcriptional R-loops. Nucleic Acids Res. 2017, 45, 10672–10692. [Google Scholar] [CrossRef] [PubMed]
- Legros, P.; Malapert, A.; Niinuma, S.; Bernard, P.; Vanoosthuyse, V. RNA processing factors Swd2.2 and Sen1 antagonize RNA Pol III-dependent transcription and the localization of condensin at Pol III genes. PLoS Genet. 2014, 10, e1004794. [Google Scholar] [CrossRef] [PubMed]
- El Hage, A.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.X.; Kobiyama, K.; Shen, Y.J.; LeBert, N.; Ahmad, S.; Khatoo, M.; Aoshi, T.; Gasser, S.; Ishii, K.J. RNA polymerase III regulates cytosolic RNA:DNA hybrids and intracellular microRNA expression. J. Biol. Chem. 2015, 290, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Becherel, O.J.; Sun, J.; Yeo, A.J.; Nayler, S.; Fogel, B.L.; Gao, F.; Coppola, G.; Criscuolo, C.; De Michele, G.; Wolvetang, E.; et al. A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum. Mol. Genet. 2015, 24, 5759–5774. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, M.; Sanjiv, K.; Helleday, T.; Herr, P.; Mortusewicz, O. The spliceosome U2 snRNP factors promote genome stability through distinct mechanisms; transcription of repair factors and R-loop processing. Oncogenesis 2016, 5, e280. [Google Scholar] [CrossRef] [PubMed]
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Mischo, H.E.; Gomez-Gonzalez, B.; Grzechnik, P.; Rondon, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzechnik, P.; Gdula, M.R.; Proudfoot, N.J. Pcf11 orchestrates transcription termination pathways in yeast. Genes Dev. 2015, 29, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic effects of topoisomerase I inhibition on R-loops and short transcripts at active promoters. PLoS ONE 2016, 11, e0147053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.; Lieber, M.R. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol. Cell. Biol. 2009, 29, 3124–3133. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Choe, J.; Seo, Y.S. The sen1+ gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 1999, 38, 14697–14710. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [PubMed]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martinez, S.; Tsanov, N.; Abou Merhi, R.; Maiorano, D. An ATR-dependent function for the ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA–DNA hybrids are required for efficient double-strand break repair. Cell 2016, 167, 1001–1013.e1007. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalez, B.; Aguilera, A. Activation-induced cytidine deaminase action is strongly stimulated by mutations of the THO complex. Proc. Natl. Acad. Sci. USA 2007, 104, 8409–8414. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Lieber, M.R. AID and reactive oxygen species can induce DNA breaks within human chromosomal translocation fragile zones. Mol. Cell 2017, 68, 901–912.e903. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.C.; Wang, S.; Ma, W.K.; Al Husini, N.; Dhoondia, Z.; Ansari, A.; Pascuzzi, P.E.; Tran, E.J. Regulated formation of lncRNA–DNA hybrids enables faster transcriptional induction and environmental adaptation. Mol. Cell 2016, 61, 393–404. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanoosthuyse, V. Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Non-Coding RNA 2018, 4, 9. https://doi.org/10.3390/ncrna4020009
Vanoosthuyse V. Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Non-Coding RNA. 2018; 4(2):9. https://doi.org/10.3390/ncrna4020009
Chicago/Turabian StyleVanoosthuyse, Vincent. 2018. "Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops" Non-Coding RNA 4, no. 2: 9. https://doi.org/10.3390/ncrna4020009
APA StyleVanoosthuyse, V. (2018). Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Non-Coding RNA, 4(2), 9. https://doi.org/10.3390/ncrna4020009