

miRinGO: Prediction of Biological Processes Indirectly Targeted by Human microRNAs


Abstract
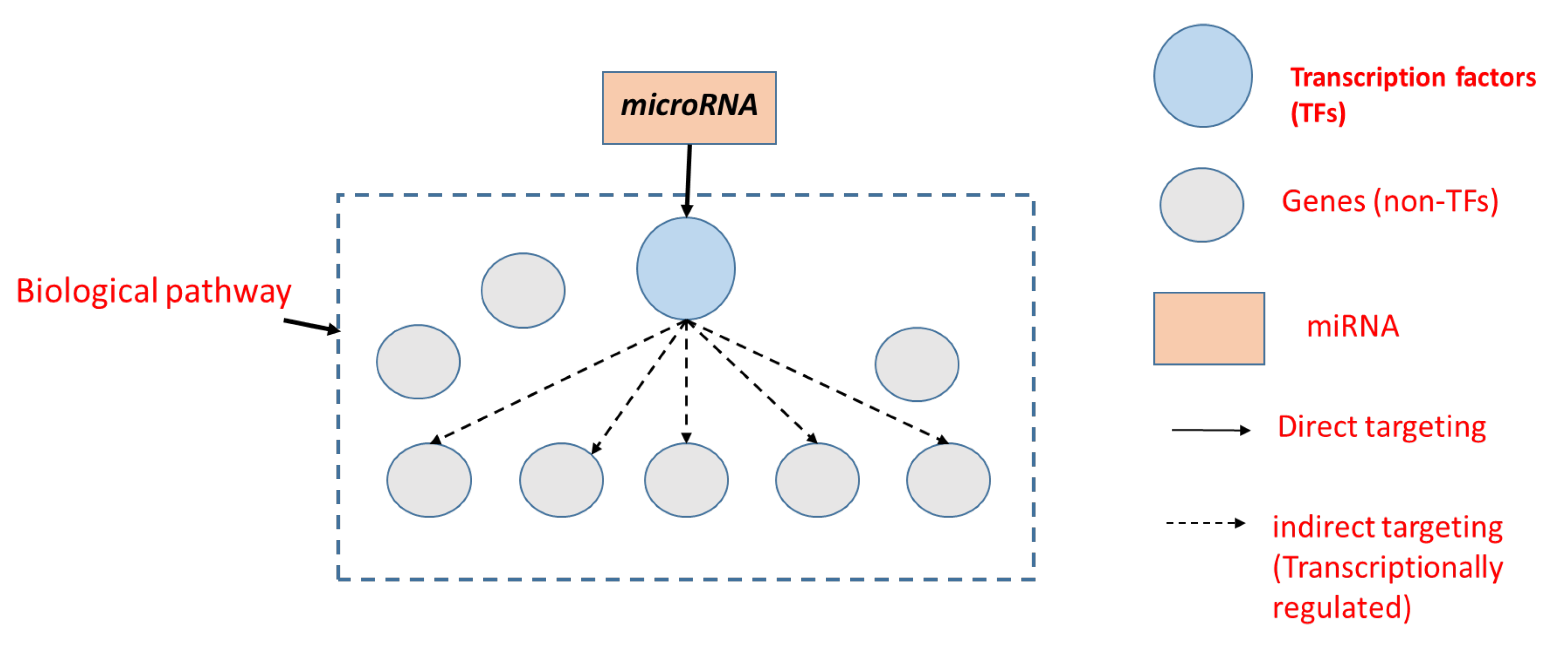
:1. Introduction
2. Methods
2.1. Overall Pipeline
2.2. Input Data
2.3. Test Dataset
3. Results
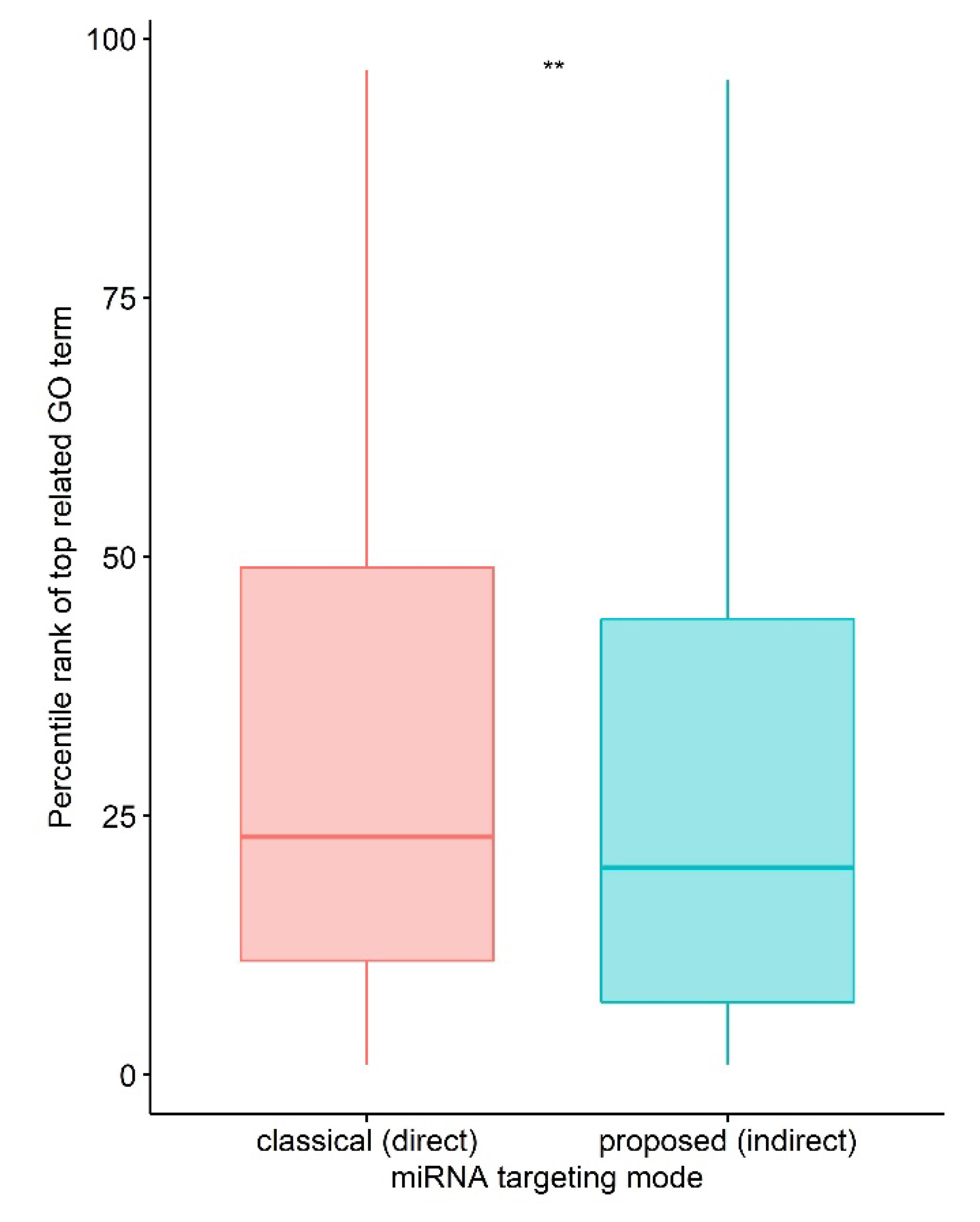
3.1. MicroRNA Indirect vs. Direct Targeting
3.2. Effect of Number of miRNA Targets
3.3. Indirect Targeting Reveals Role of miRNAs in Developmental Processes
3.4. Case Study: Role of miR-9 in Neurogenesis
3.5. Multiple miRNAs GO Analysis
3.6. R Shiny Application
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2014, 25, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomari, Y.; Zamore, P.D. Perspective: Machines for RNAi. Genes Dev. 2005, 19, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Linsley, P.S.; Schelter, J.; Burchard, J.; Kibukawa, M.; Martin, M.M.; Bartz, S.R.; Johnson, J.M.; Cummins, J.M.; Raymond, C.K.; Dai, H.; et al. Transcripts Targeted by the MicroRNA-16 Family Cooperatively Regulate Cell Cycle Progression. Mol. Cell. Biol. 2007, 27, 2240–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Gosline, S.J.; Gurtan, A.M.; JnBaptiste, C.K.; Bosson, A.; Milani, P.; Dalin, S.; Matthews, B.J.; Yap, Y.S.; Sharp, P.A.; Fraenkel, E. Elucidating MicroRNA Regulatory Networks Using Transcriptional, Post-transcriptional, and Histone Modification Measurements. Cell Rep. 2015, 14, 310–319. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Yu, Z.; Purisima, E.O.; Wang, E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006, 2, 46. [Google Scholar] [CrossRef] [Green Version]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and Local Architecture of the Mammalian microRNA–Transcription Factor Regulatory Network. PLOS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Jin, W.; Mulas, F.; Gaertner, B.; Sui, Y.; Wang, J.; Matta, I.; Zeng, C.; Vinckier, N.; Wang, A.; Nguyen-Ngoc, K.V.; et al. A network of microRNAs acts to promote cell cycle exit and differentiation of human pancreatic endocrine cells. Iscience 2019, 21, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T. DIANA-miRPath v3.0: De-ciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Preusse, M.; Theis, F.; Mueller, N.S. miTALOS v2: Analyzing Tissue Specific microRNA Function. PLoS ONE 2016, 11, e0151771. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Laurent, L.C.; Shamir, R. Towards computational prediction of microRNA function and activity. Nucleic Acids Res. 2010, 38, e160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, F.; Fehlmann, T.; Solomon, J.; Schwed, L.; Grammes, N.; Backes, C.; Van Keuren-Jensen, K.; Craig, D.W.; Meese, E.; Keller, A. miEAA 2.0: Integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res. 2020, 48, W521–W528. [Google Scholar] [CrossRef] [PubMed]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and net-work-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. Comput. Softw. J. Sci. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.-Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [Green Version]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.-C. Passing Messages between Biological Networks to Refine Predicted Interactions. PLoS ONE 2013, 8, e64832. [Google Scholar] [CrossRef]
- GTEx Consortium; Ardlie, K.G.; Deluca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J. Ensembl 2018. Nucleic Acids Res. 2017, 46, D754–D761. [Google Scholar] [CrossRef]
- Huntley, R.P.; Kramarz, B.; Sawford, T.; Umrao, Z.; Kalea, A.Z.; Acquaah, V.; Martin, M.-J.; Mayr, M.; Lovering, R.C. Expanding the horizons of microRNA bioinformatics. Rna 2018, 24, 1005–1017. [Google Scholar] [CrossRef]
- Coolen, M.; Katz, S.; Bally-Cuif, L. miR-9: A versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013, 7, 220. [Google Scholar] [CrossRef] [Green Version]
- Madelaine, R.; Sloan, S.A.; Huber, N.; Notwell, J.H.; Leung, L.C.; Skariah, G. MicroRNA-9 couples brain neurogenesis and an-giogenesis. Cell Rep. 2017, 20, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Ivey, K.N.; Srivastava, D. MicroRNAs as Regulators of Differentiation and Cell Fate Decisions. Cell Stem Cell 2010, 7, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Haga, C.L.; Phinney, D.G. MicroRNAs in the imprinted DLK1-DIO3 region repress the epithelial-to-mesenchymal transi-tion by targeting the TWIST1 protein signaling network. J. Biol. Chem. 2012, 287, 42695–42707. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Qian, P.; Zhang, X.; Zhang, M.; Wang, H.; Wu, M. Autocrine/paracrine human growth hormone-stimulated Mi-croRNA 96-182-183 cluster promotes epithelial-mesenchymal transition and invasion in breast cancer. J. Biol. Chem. 2015, 290, 13812–13829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinzón, N.; Li, B.; Martinez, L.; Sergeeva, A.; Presumey, J.; Apparailly, F.; Seitz, H. microRNA target prediction programs predict many false positives. Genome Res. 2016, 27, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.L.; Ohtsuka, T.; González, A.; Kageyama, R. Micro RNA 9 regulates neural stem cell differentiation by controlling H es1 expression dynamics in the developing brain. Genes Cells 2012, 17, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I. DIANA-TarBase v8: A dec-ade-long collection of experimentally supported miRNA–gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features/Tools | mirPath v3.0 | StarBase | miTALOS | miRWalk v3.0 |
|---|---|---|---|---|
| Predicted targets databases | TargetScan (v6)/microT-CDS (v5.0) | TargetScan/miRanda/PITA/RNA22/PicTar/… | TargetScan/miRanda | TargetScan (v7.1)/miRDB |
| Validated targets databases | TarBase v7.0 | CLIP-Seq data | CLIP-Seq data | miRTarbase |
| Pathways/GO terms databases | KEGG/GO categories | KEGG/GO/Reactome/BioCarta | KEGG/WikiPathways/Reactome | KEGG/GO/Reactome |
| Inclusion of indirect targets? | No | No | No | No |
| Tissue specific? | No | No | Yes | No |
| Allows multiple miRNAs? | Yes | No | Yes | Yes |
| GO Term ID | GO Term | Number of TFs | Number of Genes | Parent Process |
|---|---|---|---|---|
| GO:0001714 | endodermal cell fate specification | 5 | 5 | developmental process |
| GO:0003211 | cardiac ventricle formation | 5 | 5 | developmental process |
| GO:0003357 | noradrenergic neuron differentiation | 5 | 5 | developmental process |
| GO:0021520 | spinal cord motor neuron cell fate specification | 7 | 7 | developmental process |
| GO:0021902 | commitment of neuronal cell to specific neuron type in forebrain | 7 | 7 | developmental process |
| Tool | Highest Ranking GO Term Related to Neurogenesis | Rank |
|---|---|---|
| miRinGO | Nervous system development | 1 |
| mirPath v3 | Regulation of neuron maturation | 11 |
| miRWalk v3 | Axonogenesis | 13 |
| StarBase v3 | Neurogenesis | 23 |
| miTALOS v2 | N/A | N/A |
| miRNAs | Rank of Top GO Term Related to EMT |
|---|---|
| miR-200a/miR-141 | 132 |
| miR-200b/miR-200c/miR-429 | 147 |
| miR-205-5p | 105 |
| All three miRNAs | 70 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayed, M.; Park, J.W. miRinGO: Prediction of Biological Processes Indirectly Targeted by Human microRNAs. Non-Coding RNA 2023, 9, 11. https://doi.org/10.3390/ncrna9010011
Sayed M, Park JW. miRinGO: Prediction of Biological Processes Indirectly Targeted by Human microRNAs. Non-Coding RNA. 2023; 9(1):11. https://doi.org/10.3390/ncrna9010011
Chicago/Turabian StyleSayed, Mohammed, and Juw Won Park. 2023. "miRinGO: Prediction of Biological Processes Indirectly Targeted by Human microRNAs" Non-Coding RNA 9, no. 1: 11. https://doi.org/10.3390/ncrna9010011
APA StyleSayed, M., & Park, J. W. (2023). miRinGO: Prediction of Biological Processes Indirectly Targeted by Human microRNAs. Non-Coding RNA, 9(1), 11. https://doi.org/10.3390/ncrna9010011