
Non-Coding RNA-Dependent Regulation of Mitochondrial Dynamics in Cancer Pathophysiology


Abstract
:1. Introduction
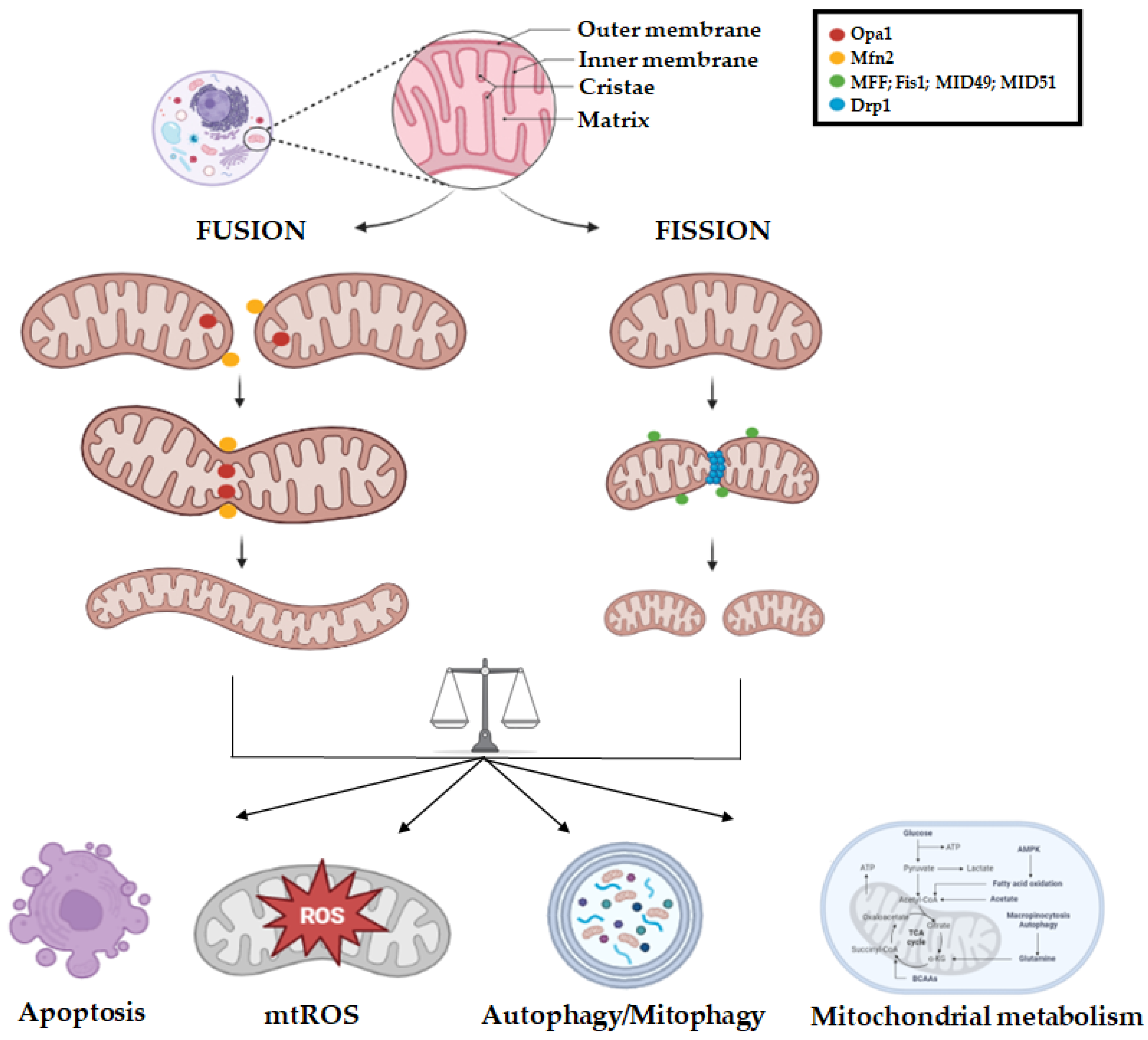
2. Mitochondrial Dynamics Biology
3. Mitochondrial Dynamics Aberrations in Cancer
4. Non-Coding RNA-Regulation of Mitochondrial Dynamics
4.1. Non-Coding RNA Classification and Function
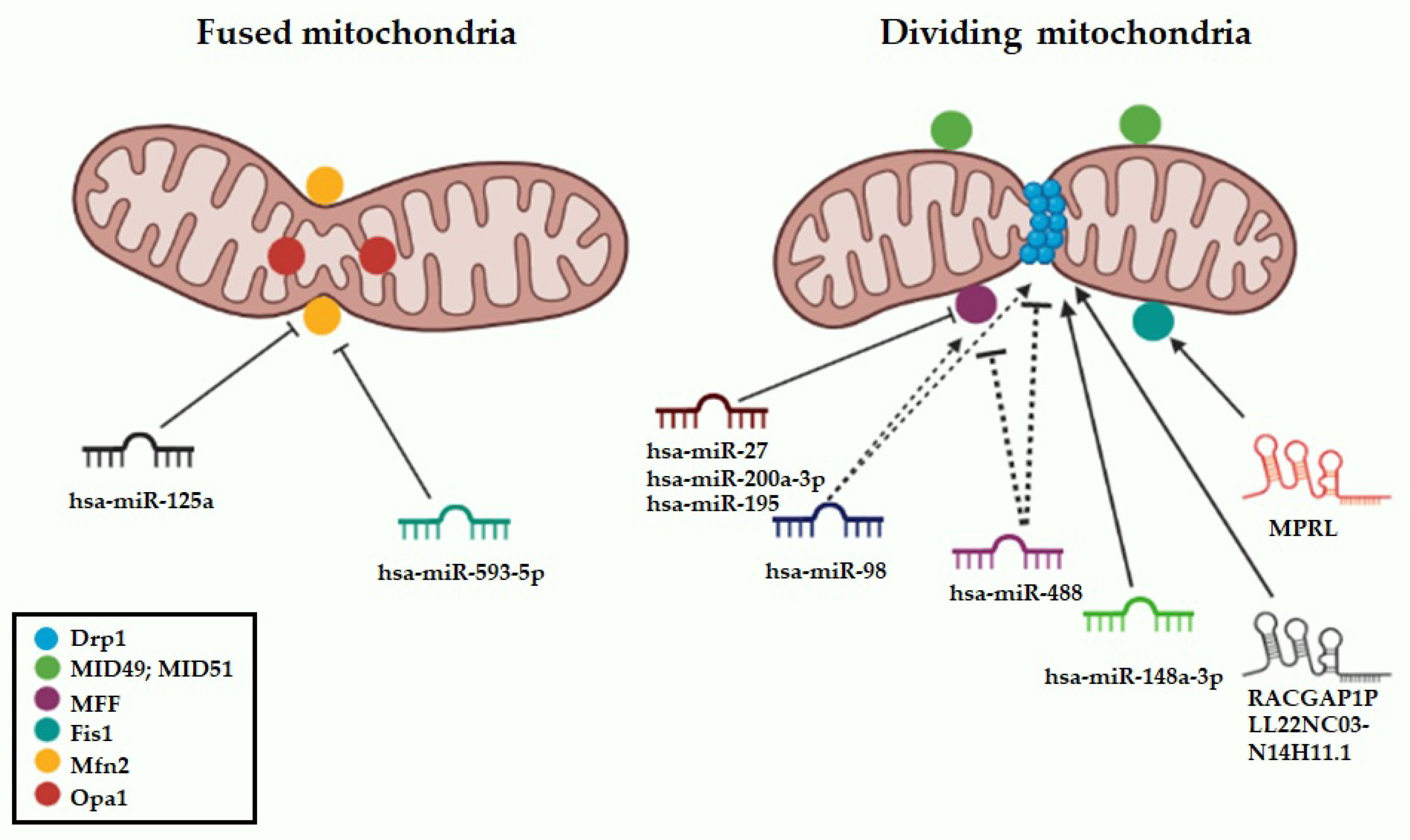
4.2. miRNAs Targeting Mitochondrial Dynamics Effectors
4.2.1. hsa-miR-125a
4.2.2. hsa-miR-148a-3p
4.2.3. hsa-miR-98
4.2.4. hsa-miR-29c-3p
4.2.5. hsa-miR-593-5p
4.2.6. has-miR-27 and hsa-miR-200-3p
4.2.7. hsa-miR-488
4.2.8. hsa-miR-195
4.3. lncRNAs Targeting Mitochondrial Dynamics Effectors
4.3.1. RACGAP1P
4.3.2. MPRL
4.3.3. LL22NC03-N14H11.1
4.3.4. CDK6-AS1
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, L.; Goel, A.; Wang, X. Novel paradigms of mitochondrial biology and function: Potential clinical significance in the era of precision medicine. Cell Biol. Toxicol. 2022, 38, 371–375. [Google Scholar] [CrossRef]
- Schirrmacher, V. Mitochondria at Work: New Insights into Regulation and Dysregulation of Cellular Energy Supply and Metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Abd Radzak, S.M.; Mohd Khair, S.Z.; Ahmad, F.; Patar, A.; Idris, Z.; Mohamed Yusoff, A. Insights regarding mitochondrial DNA copy number alterations in human cancer (Review). Int. J. Mol. Med. 2022, 50, 104. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Park, J.Y.; Lee, H.; Song, E.S.; Kuk, M.U.; Joo, J.; Oh, S.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells 2021, 10, 3003. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial adaptation in cancer drug resistance: Prevalence, mechanisms, and management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Vera, J.; Wolkenhauer, O. The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef]
- Lee, H.; Smith, S.B.; Yoon, Y. The short variant of the mitochondrial dynamin OPA1 maintains mitochondrial energetics and cristae structure. J. Biol. Chem. 2017, 292, 7115–7130. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; van der Bliek, A.M. The Novel Tail-anchored Membrane Protein Mff Controls Mitochondrial and Peroxisomal Fission in Mammalian Cells. Mol. Biol. Cell 2008, 19, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.-H.; Cao, Y.-T.; Zhang, L.-L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basañez, G.; Meda, P.; et al. Membrane Remodeling Induced by the Dynamin-Related Protein Drp1 Stimulates Bax Oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Wan, Y.-Y.; Zhang, J.-F.; Yang, Z.-J.; Jiang, L.-P.; Wei, Y.-F.; Lai, Q.-N.; Wang, J.-B.; Xin, H.-B.; Han, X.-J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Moro, L. Mitochondrial Dysfunction in Aging and Cancer. J. Clin. Med. 2019, 8, 1983. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochim. Et Biophys. Acta BBA Rev. Cancer 2013, 1836, 336–344. [Google Scholar] [CrossRef] [Green Version]
- Errichiello, E.; Venesio, T. Mitochondrial DNA variants in colorectal carcinogenesis: Drivers or passengers? J. Cancer Res. Clin. Oncol. 2017, 143, 1905–1914. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Jiang, L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Aquila, P.; Ronchetti, D.; Gallo Cantafio, M.E.; Todoerti, K.; Taiana, E.; Fabiani, F.; Montesanto, A.; Neri, A.; Passarino, G.; Viglietto, G.; et al. Epigenetic Regulation of Mitochondrial Quality Control Genes in Multiple Myeloma: A Sequenom MassARRAY Pilot Investigation on HMCLs. J. Clin. Med. 2021, 10, 1295. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Halilovic, E.; Solit, D.B. Therapeutic strategies for inhibiting oncogenic BRAF signaling. Curr. Opin. Pharm. 2008, 8, 419–426. [Google Scholar] [CrossRef]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraz, L.S.; da Costa, R.T.; da Costa, C.A.; Ribeiro, C.A.J.; Arruda, D.C.; Maria-Engler, S.S.; Rodrigues, T. Targeting mitochondria in melanoma: Interplay between MAPK signaling pathway and mitochondrial dynamics. Biochem. Pharm. 2020, 178, 114104. [Google Scholar] [CrossRef]
- Banerjee, R.; Mukherjee, A.; Nagotu, S. Mitochondrial dynamics and its impact on human health and diseases: Inside the DRP1 blackbox. J. Mol. Med. 2022, 100, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Dasgupta, A.; Chen, K.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Agarwal, E.; Chae, Y.C.; Lee, Y.G.; Garlick, D.S.; Storaci, A.M.; Ferrero, S.; Gaudioso, G.; Gianelli, U.; Vaira, V.; et al. Mitochondrial fission factor is a novel Myc-dependent regulator of mitochondrial permeability in cancer. EBioMedicine 2019, 48, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Chae, Y.C.; Kossenkov, A.V.; Lee, Y.G.; Tang, H.-Y.; Agarwal, E.; Gabrilovich, D.I.; Languino, L.R.; Speicher, D.W.; Shastrula, P.K.; et al. MFF Regulation of Mitochondrial Cell Death Is a Therapeutic Target in Cancer. Cancer Res. 2019, 79, 6215–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herkenne, S.; Ek, O.; Zamberlan, M.; Pellattiero, A.; Chergova, M.; Chivite, I.; Novotná, E.; Rigoni, G.; Fonseca, T.B.; Samardzic, D.; et al. Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab. 2020, 31, 987–1003.e8. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells 2020, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome c Release and Apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Carmona, C.A.; Dalla Pozza, E.; Ambrosini, G.; Cisterna, B.; Palmieri, M.; Decimo, I.; Cuezva, J.M.; Bottani, E.; Dando, I. Mitochondrial Elongation and OPA1 Play Crucial Roles during the Stemness Acquisition Process in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 3432. [Google Scholar] [CrossRef]
- Zamberlan, M.; Boeckx, A.; Muller, F.; Vinelli, F.; Ek, O.; Vianello, C.; Coart, E.; Shibata, K.; Christian, A.; Grespi, F.; et al. Inhibition of the mitochondrial protein Opa1 curtails breast cancer growth. J. Exp. Clin. Cancer Res. 2022, 41, 95. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 Is Required for Cisplatin-induced Processing of the Mitochondrial Fusion Protein L-Opa1 That Is Mediated by the Mitochondrial Metallopeptidase Oma1 in Gynecologic Cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorile, A.; de Rasmo, D.; Cormio, A.; Musicco, C.; Rossi, R.; Fortarezza, F.; Palese, L.; Loizzi, V.; Resta, L.; Scillitani, G.; et al. Human Ovarian Cancer Tissue Exhibits Increase of Mitochondrial Biogenesis and Cristae Remodeling. Cancers 2019, 11, 1350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, R.; Chen, R.; Liu, J. Altered Mitochondrial Dynamics, Biogenesis, and Functions in the Paclitaxel-Resistant Lung Adenocarcinoma Cell Line A549/Taxol. Med. Sci. Monit. 2020, 26, e918216-1. [Google Scholar] [CrossRef]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 4, e126915. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta BBA Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Vendramin, R.; Marine, J.; Leucci, E. Non-coding RNAs: The dark side of nuclear–mitochondrial communication. EMBO J. 2017, 36, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Gusic, M.; Prokisch, H. ncRNAs: New Players in Mitochondrial Health and Disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Amodio, N.; di Martino, M.T.; Neri, A.; Tagliaferri, P.; Tassone, P. Non-coding RNA: A novel opportunity for the personalized treatment of multiple myeloma. Expert Opin. Biol. 2013, 13, S125–S137. [Google Scholar] [CrossRef]
- Morelli, E.; Gullà, A.; Rocca, R.; Federico, C.; Raimondi, L.; Malvestiti, S.; Agosti, V.; Rossi, M.; Costa, G.; Giavaresi, G.; et al. The Non-Coding RNA Landscape of Plasma Cell Dyscrasias. Cancers 2020, 12, 320. [Google Scholar] [CrossRef] [Green Version]
- Purohit, P.K.; Edwards, R.; Tokatlidis, K.; Saini, N. MiR-195 regulates mitochondrial function by targeting mitofusin-2 in breast cancer cells. RNA Biol. 2019, 16, 918–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; et al. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Bharat; Dogra, N.; Singh, S. Small Regulatory Molecules Acting Big in Cancer: Potential Role of Mito-miRs in Cancer. Curr. Mol. Med. 2019, 19, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, G.C.; Magalhães, L.; Ribeiro-dos-Santos, Â.; Vidal, A.F. Mitochondrial Epigenetics: Non-Coding RNAs as a Novel Layer of Complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef] [Green Version]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. In microRNA: Medical Evidence: From Molecular Biology to Clinical Practice; Springer: Berlin/Heidelberg, Germany, 2015; Volume 888, pp. 123–154. [Google Scholar]
- Sanchez Calle, A.; Kawamura, Y.; Yamamoto, Y.; Takeshita, F.; Ochiya, T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018, 109, 2093–2100. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yoshitomi, T.; Hu, J.-F.; Cui, J. Long noncoding RNAs coordinate functions between mitochondria and the nucleus. Epigenet. Chromatin 2017, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- de Paepe, B.; Lefever, S.; Mestdagh, P. How long noncoding RNAs enforce their will on mitochondrial activity: Regulation of mitochondrial respiration, reactive oxygen species production, apoptosis, and metabolic reprogramming in cancer. Curr. Genet. 2018, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, L.; Li, H.; Sun, T.; Wen, X.; Li, X.; Meng, Y.; Li, Y.; Liu, M.; Liu, S.; et al. Nuclear-Encoded lncRNA MALAT1 Epigenetically Controls Metabolic Reprogramming in HCC Cells through the Mitophagy Pathway. Mol. Nucleic Acids 2021, 23, 264–276. [Google Scholar] [CrossRef]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int. J. Oncol. 2018, 53, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, W.; Li, Z.; Chen, Z.; Zhi, X.; Xu, J.; Li, Q.; Wang, L.; Huang, X.; Wang, L.; et al. MicroRNA-148a-3p enhances cisplatin cytotoxicity in gastric cancer through mitochondrial fission induction and cyto-protective autophagy suppression. Cancer Lett. 2017, 410, 212–227. [Google Scholar] [CrossRef]
- Luan, T.; Fu, S.; Huang, L.; Zuo, Y.; Ding, M.; Li, N.; Chen, J.; Wang, H.; Wang, J. MicroRNA-98 promotes drug resistance and regulates mitochondrial dynamics by targeting LASS2 in bladder cancer cells. Exp. Cell Res. 2018, 373, 188–197. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Jin, K.; Dong, R.; Gao, C.; Si, L.; Feng, Z.; Zhang, H.; Tian, H. Negatively Regulated by miR-29c-3p, MTFR1 Promotes the Progression and Glycolysis in Lung Adenocarcinoma via the AMPK/mTOR Signalling Pathway. Front. Cell Dev. Biol. 2021, 9, 771824. [Google Scholar] [CrossRef]
- Fan, S.; Liu, B.; Sun, L.; Lv, X.; Lin, Z.; Chen, W.; Chen, W.; Tang, Q.; Wang, Y.; Su, Y.; et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p–MFF axis. Oncotarget 2015, 6, 14885–14904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, H.; Kim, J.; Jayabalan, A.K.; Lee, H.; Kang, H.; Cho, D.-H.; Ohn, T.; Nam, S.W.; Kim, W.; Lee, E.K. miR-27 regulates mitochondrial networks by directly targeting the mitochondrial fission factor. Exp. Mol. Med. 2014, 46, e123. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Tak, H.; Park, S.J.; Jo, Y.K.; Cho, D.H.; Lee, E.K. microRNA-200a-3p enhances mitochondrial elongation by targeting mitochondrial fission factor. BMB Rep. 2017, 50, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, H.; Eun, J.W.; Kim, J.; Park, S.J.; Kim, C.; Ji, E.; Lee, H.; Kang, H.; Cho, D.-H.; Lee, K.; et al. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ. 2017, 24, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, H.; Kang, H.; Ji, E.; Hong, Y.; Kim, W.; Lee, E.K. Potential use of TIA-1, MFF, microRNA-200a-3p, and microRNA-27 as a novel marker for hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 497, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Feng, Z.; Gu, J.; Li, X.; Dong, Q.; Liu, K.; Li, Y.; OuYang, L. microRNA-488 inhibits chemoresistance of ovarian cancer cells by targeting Six1 and mitochondrial function. Oncotarget 2017, 8, 80981–80993. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Ren, K.; Wang, M.; Wang, J.; Li, E.; Hou, C.; Su, Y.; Jin, Y.; Zou, Q.; Zhou, P.; et al. Long non-coding RNA RACGAP1P promotes breast cancer invasion and metastasis via miR-345-5p/RACGAP1-mediated mitochondrial fission. Mol. Oncol. 2021, 15, 543–559. [Google Scholar] [CrossRef]
- Tian, T.; Lv, X.; Pan, G.; Lu, Y.; Chen, W.; He, W.; Lei, X.; Zhang, H.; Liu, M.; Sun, S.; et al. Long Noncoding RNA MPRL Promotes Mitochondrial Fission and Cisplatin Chemosensitivity via Disruption of Pre-miRNA Processing. Clin. Cancer Res. 2019, 25, 3673–3688. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.; Luo, H.; Qin, F.; Jiang, Q.; He, S.; Wang, T.; Su, J.; Song, S.; Qin, X.; Qin, Y.; et al. LncRNA LL22NC03-N14H11.1 promoted hepatocellular carcinoma progression through activating MAPK pathway to induce mitochondrial fission. Cell Death Dis. 2020, 11, 832. [Google Scholar] [CrossRef]
- Porcù, E.; Benetton, M.; Bisio, V.; da Ros, A.; Tregnago, C.; Borella, G.; Zanon, C.; Bordi, M.; Germano, G.; Manni, S.; et al. The long non-coding RNA CDK6-AS1 overexpression impacts on acute myeloid leukemia differentiation and mitochondrial dynamics. iScience 2021, 24, 103350. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Yu, M.; Gao, G.; Liang, H.; Zhou, X.; Zhu, Z.; Zhang, C.; Wang, Y.; Chen, X. MiR-125a-5p functions as a tumour suppressor in breast cancer by downregulating BAP1. J. Cell Biochem. 2018, 119, 8773–8783. [Google Scholar] [CrossRef]
- Nishida, N.; Mimori, K.; Fabbri, M.; Yokobori, T.; Sudo, T.; Tanaka, F.; Shibata, K.; Ishii, H.; Doki, Y.; Mori, M. MicroRNA-125a-5p Is an Independent Prognostic Factor in Gastric Cancer and Inhibits the Proliferation of Human Gastric Cancer Cells in Combination with Trastuzumab. Clin. Cancer Res. 2011, 17, 2725–2733. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Huang, Q.; Zhang, S.; Zhang, Q.; Chang, J.; Qiu, X.; Wang, E. Hsa-miR-125a-3p and hsa-miR-125a-5p are downregulated in non-small cell lung cancer and have inverse effects on invasion and migration of lung cancer cells. BMC Cancer 2010, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Cowden Dahl, K.D.; Dahl, R.; Kruichak, J.N.; Hudson, L.G. The Epidermal Growth Factor Receptor Responsive miR-125a Represses Mesenchymal Morphology in Ovarian Cancer Cells. Neoplasia 2009, 11, 1208-IN21. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Cuatrecasas, M.; Balaguer, F.; Hur, K.; Toiyama, Y.; Castells, A.; Boland, C.R.; Goel, A. The Clinical Significance of MiR-148a as a Predictive Biomarker in Patients with Advanced Colorectal Cancer. PLoS ONE 2012, 7, e46684. [Google Scholar] [CrossRef] [Green Version]
- Hanoun, N.; Delpu, Y.; Suriawinata, A.A.; Bournet, B.; Bureau, C.; Selves, J.; Tsongalis, G.J.; Dufresne, M.; Buscail, L.; Cordelier, P.; et al. The Silencing of MicroRNA 148a Production by DNA Hypermethylation Is an Early Event in Pancreatic Carcinogenesis. Clin. Chem. 2010, 56, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Huang, S.; He, R.; Rong, M.; Dang, Y.; Chen, G. Decreased expression and clinical significance of miR-148a in hepatocellular carcinoma tissues. Eur. J. Med. Res. 2014, 19, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Liang, L.; Wang, C.; Huang, S.; Cao, X.; Zha, R.; Liu, L.; Jia, D.; Tian, Q.; Wu, J.; et al. MicroRNA-148a Suppresses Tumor Cell Invasion and Metastasis by Downregulating ROCK1 in Gastric Cancer. Clin. Cancer Res. 2011, 17, 7574–7583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.-Q.; Yin, J.-Y.; Tang, Q.; Liu, F.-Q.; Qian, J.; Lin, J.; Shao, R.; Zhang, M.; He, L. Over-expression of miR-98 in FFPE tissues might serve as a valuable source for biomarker discovery in breast cancer patients. Int. J. Clin. Exp. Pathol. 2014, 7, 1166–1171. [Google Scholar] [PubMed]
- Hebert, C.; Norris, K.; Scheper, M.A.; Nikitakis, N.; Sauk, J.J. High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 2007, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ni, R.; Huang, Y. miR-98 targets ITGB3 to inhibit proliferation, migration, and invasion of non-small-cell lung cancer. Onco Targets 2015, 8, 2689–2697. [Google Scholar] [CrossRef] [Green Version]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, N.; Rossi, M.; Raimondi, L.; Pitari, M.R.; Botta, C.; Tagliaferri, P.; Tassone, P. miR-29s: A family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget 2015, 6, 12837–12861. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhang, W.; Wu, Z.; Liu, Y.; Shi, Y.; Gong, J.; Shen, W.; Liu, C. miR-29c-3p regulates DNMT3B and LATS1 methylation to inhibit tumor progression in hepatocellular carcinoma. Cell Death Dis. 2019, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Cai, M.; Zhang, Y.; Tao, L.; Guo, R. miR-29c-3p inhibits autophagy and cisplatin resistance in ovarian cancer by regulating FOXP1/ATG14 pathway. Cell Cycle 2020, 19, 193–206. [Google Scholar] [CrossRef]
- Zou, T.; Gao, Y.; Qie, M. MiR-29c-3p inhibits epithelial-mesenchymal transition to inhibit the proliferation, invasion and metastasis of cervical cancer cells by targeting SPARC. Ann. Transl. Med. 2021, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Luo, S.; Ji, C.; Shi, J. miR-29c-3p regulates proliferation and migration in ovarian cancer by targeting KIF4A. World J. Surg. Oncol. 2020, 18, 315. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Zhang, Y.; Li, K.; Wang, M.; Li, J.; Qi, Z.; Wu, J.; Wang, Z.; Ling, L.; Liu, H.; et al. miR-593-5p inhibit cell proliferation by targeting PLK1 in non small cell lung cancer cells. Pathol. Res. Pract. 2020, 216, 152786. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Shen, B.; Ma, Z.-C.; Xu, Y.-Y.; Lou, Y.-L.; Chen, M. MiR-593-5p inhibited proliferation and migration of lung adenocarcinoma by targeting ICAM-1. Eur. Rev. Med. Pharm. Sci 2020, 24, 4298–4305. [Google Scholar]
- Tan, T.; Xu, X.-H.; Lu, X.-H.; Wang, X.-W. MiRNA-200a-3p suppresses the proliferation, migration and invasion of non-small cell lung cancer through targeting IRS2. Eur. Rev. Med. Pharm. Sci. 2020, 24, 712–720. [Google Scholar]
- Zhang, J.; Cao, Z.; Yang, G.; You, L.; Zhang, T.; Zhao, Y. MicroRNA-27a (miR-27a) in Solid Tumors: A Review Based on Mechanisms and Clinical Observations. Front. Oncol. 2019, 9, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.; Zhang, L.; Chen, X.; Chen, S.; Shi, Y.; Li, J. Silencing microRNA-27a inhibits proliferation and invasion of human osteosarcoma cells through the SFRP1-dependent Wnt/β-catenin signaling pathway. Biosci. Rep. 2019, 39, BSR20182366. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Chen, Y.-X.; Wu, N.-Y.; Yin, J.-Y.; Li, X.-P.; Huang, H.-S.; Zhang, W.; Zhou, H.-H.; Liu, Z.-Q. MiR-488 inhibits proliferation and cisplatin sensibility in non-small-cell lung cancer (NSCLC) cells by activating the eIF3a-mediated NER signaling pathway. Sci. Rep. 2017, 7, 40384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Lu, G.; Ke, X.; Lu, X.; Wang, X.; Li, H.; Ren, M.; He, S. miR-488 acts as a tumor suppressor gene in gastric cancer. Tumor Biol. 2016, 37, 8691–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.-Y.; Wang, X.-Q.; Sun, L.-F. MicroRNA-488 inhibits ovarian cancer cell metastasis through regulating CCNG1 and p53 expression. Eur. Rev. Med. Pharm. Sci. 2020, 24, 2902–2910. [Google Scholar]
- Li, J.; Fan, R.; Xiao, H. Circ_ZFR contributes to the paclitaxel resistance and progression of non-small cell lung cancer by upregulating KPNA4 through sponging miR-195-5p. Cancer Cell Int. 2021, 21, 15. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xia, T.; Cao, M.; Zhang, P.; Shi, G.; Chen, L.; Zhang, J.; Yin, J.; Wu, P.; Cai, B.; et al. LncRNA BANCR Promotes Pancreatic Cancer Tumorigenesis via Modulating MiR-195-5p/Wnt/β-Catenin Signaling Pathway. Technol. Cancer Res. Treat. 2019, 18, 153303381988796. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, Y.; Liu, C.; Chen, X.; Qi, Y.; Jiang, Y.; Zou, C.; Zhang, X.; Liu, S.; Wang, X.; et al. Analysis of MiR-195 and MiR-497 Expression, Regulation and Role in Breast Cancer. Clin. Cancer Res. 2011, 17, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Menigatti, M.; Staiano, T.; Manser, C.N.; Bauerfeind, P.; Komljenovic, A.; Robinson, M.; Jiricny, J.; Buffoli, F.; Marra, G. Epigenetic silencing of monoallelically methylated miRNA loci in precancerous colorectal lesions. Oncogenesis 2013, 2, e56. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-Y.; Chen, D.-P.; Qi, B.; Li, M.-Y.; Zhu, Y.-Y.; Yin, W.-J.; He, L.; Yu, Y.; Li, Z.-Y.; Lin, L.; et al. Pseudogene RACGAP1P activates RACGAP1/Rho/ERK signalling axis as a competing endogenous RNA to promote hepatocellular carcinoma early recurrence. Cell Death Dis. 2019, 10, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydarnezhad Asl, M.; Pasban Khelejani, F.; Bahojb Mahdavi, S.Z.; Emrahi, L.; Jebelli, A.; Mokhtarzadeh, A. The various regulatory functions of long noncoding RNAs in apoptosis, cell cycle, and cellular senescence. J. Cell. Biochem. 2022, 123, 995–1024. [Google Scholar] [CrossRef]
- Quinn, J.J.; Zhang, Q.C.; Georgiev, P.; Ilik, I.A.; Akhtar, A.; Chang, H.Y. Rapid evolutionary turnover underlies conserved lncRNA–genome interactions. Genes Dev. 2016, 30, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Fernando, T.R.; Rodriguez-Malave, N.I.; Waters, E.V.; Yan, W.; Casero, D.; Basso, G.; Pigazzi, M.; Rao, D.S. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol. Cancer Res. 2015, 13, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug. Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gullà, A.; et al. Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood 2018, 132, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; de Luca, A.; Giavaresi, G.; Raimondo, S.; Gallo, A.; Taiana, E.; Alessandro, R.; Rossi, M.; Neri, A.; Viglietto, G.; et al. Non-Coding RNAs in Multiple Myeloma Bone Disease Pathophysiology. Noncoding RNA 2020, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Amodio, N.; Martino, M.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. From Target Therapy to miRNA Therapeutics of Human Multiple Myeloma: Theoretical and Technological Issues in the Evolving Scenario. Curr. Drug Targets 2013, 14, 1144–1149. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Non-Coding RNA | Cancer Type/ Expression | Mitochondrial Dynamics Target | Phenotypic Effects | References |
|---|---|---|---|---|
| hsa-miR-125a | Downregulated in pancreatic cancer | MFN2 | miR-125a decreases cancer cell survival and migration | [65] |
| hsa-miR-148a-3p | Downregulated in gastric cancer | AKAP1 | miR-148a-3p inhibits gastric cancer cell growth, induces mitochondrial apoptosis, and increases cisplatin sensitivity | [66] |
| hsa-miR-98 | Upregulated in bladder cancer cells | LASS2 | miR-98 promotes cancer cell proliferation, inhibits apoptosis, and induces multidrug resistance | [67] |
| hsa-miR-29c-3p | Downregulated in lung adenocarcinoma | MTFR1 | miR-29c-3p suppresses proliferation, migration, and invasion of lung cancer cells | [68] |
| hsa-miR-593-5p | Downregulated in tongue squamous cell carcinoma after cisplatin | MFF | miR-593-5p inhibits apoptotic cell death and decreases cisplatin sensitivity | [69] |
| hsa-miR-27 and hsa-miR-200a-3p | Downregulated in several tumor types | TIA-1, MFF | miR-27 and miR-200a-3p inhibit migration and invasion of cancer cells | [70,71,72,73] |
| hsa-miR-488 | Downregulated in ovarian cancer | Six-1 | miR-488 inhibits ovarian cancer cell proliferation, induces apoptosis, and increases cisplatin sensitivity | [74] |
| hsa-miR-195 | Downregulated in breast cancer | MFN2 | miR-195 exerts pro-apoptotic effects in breast cancer cells | [55] |
| RACGAP1P | Upregulated in breast cancer | Drp1 | RACGAP1P promotes breast cancer cell proliferation and metastatic behaviour | [75] |
| MPRL | Downregulated in tongue squamous cell carcinoma | Fis1 | MPRL inhibits tumor growth in TSCC cells, activates apoptosis, and induces cisplatin sensitivity | [76] |
| LL22NC03-N14H11.1 | Upregulated in hepatocarcinoma | Drp1 | LL22NC03-N14H11.1 promotes cell survival, migration, and invasion of HCC cells | [77] |
| CDK6-AS1 | Upregulated in acute myeloid leukemia | − | CDK6-AS1 inhibits myeloid differentiation, and increases stemness, drug resistance, and tumor aggressiveness | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo Cantafio, M.E.; Torcasio, R.; Viglietto, G.; Amodio, N. Non-Coding RNA-Dependent Regulation of Mitochondrial Dynamics in Cancer Pathophysiology. Non-Coding RNA 2023, 9, 16. https://doi.org/10.3390/ncrna9010016
Gallo Cantafio ME, Torcasio R, Viglietto G, Amodio N. Non-Coding RNA-Dependent Regulation of Mitochondrial Dynamics in Cancer Pathophysiology. Non-Coding RNA. 2023; 9(1):16. https://doi.org/10.3390/ncrna9010016
Chicago/Turabian StyleGallo Cantafio, Maria Eugenia, Roberta Torcasio, Giuseppe Viglietto, and Nicola Amodio. 2023. "Non-Coding RNA-Dependent Regulation of Mitochondrial Dynamics in Cancer Pathophysiology" Non-Coding RNA 9, no. 1: 16. https://doi.org/10.3390/ncrna9010016
APA StyleGallo Cantafio, M. E., Torcasio, R., Viglietto, G., & Amodio, N. (2023). Non-Coding RNA-Dependent Regulation of Mitochondrial Dynamics in Cancer Pathophysiology. Non-Coding RNA, 9(1), 16. https://doi.org/10.3390/ncrna9010016