Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
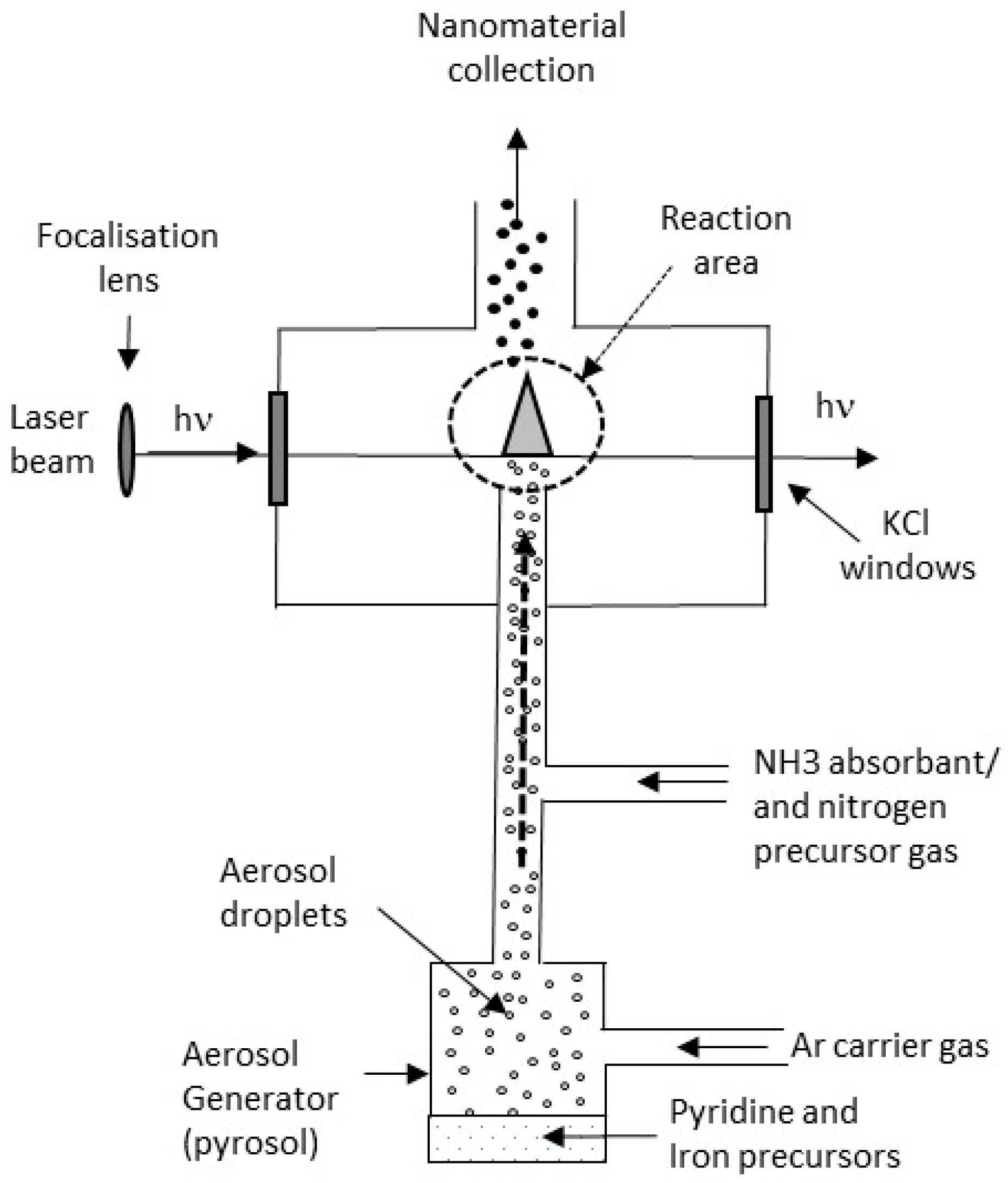
2.1. Synthesis
2.2. Characterization
2.3. Electrode Preparation and Electrochemical Measurement
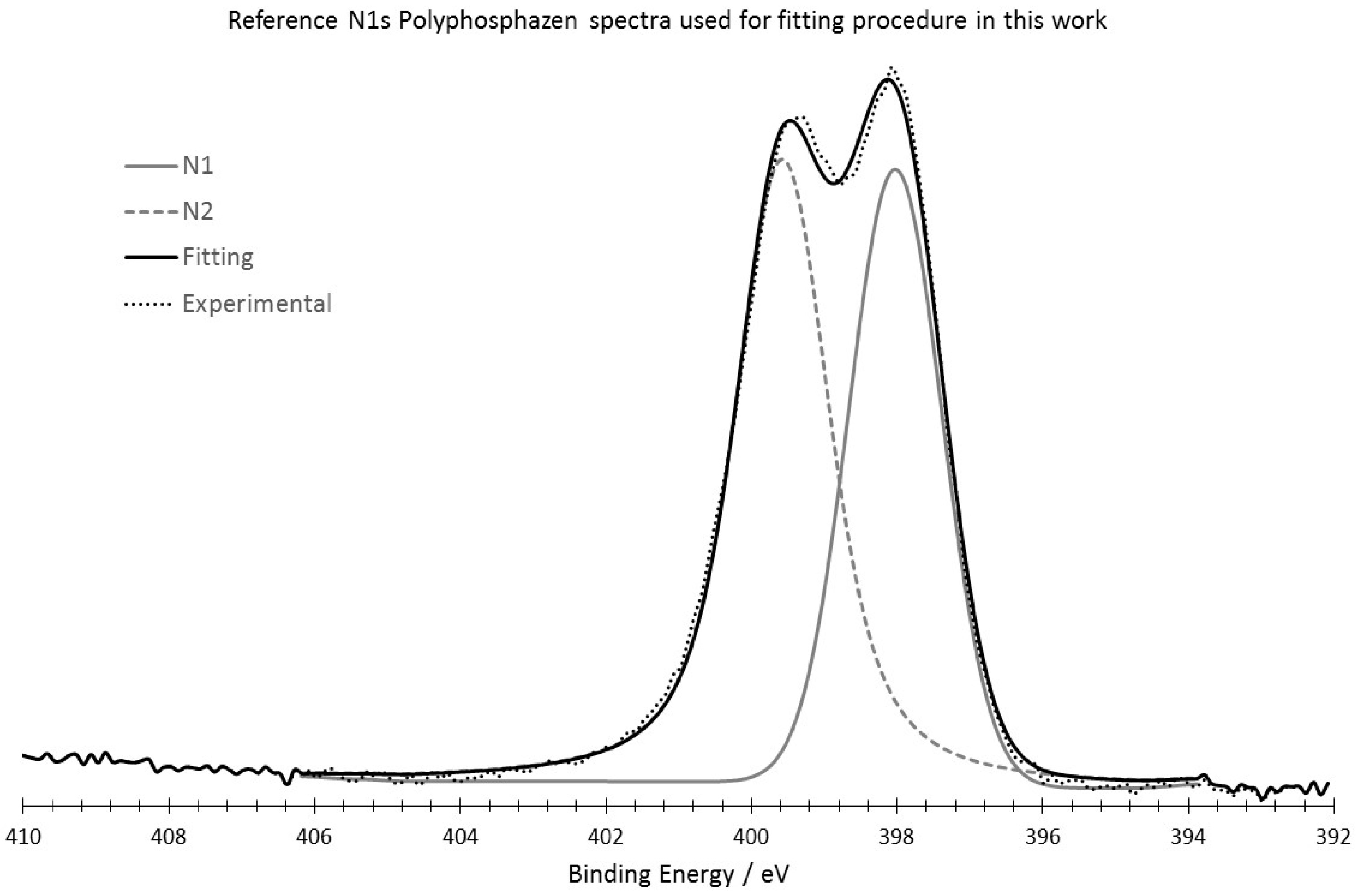
2.4. XPS Analysis
3. Results
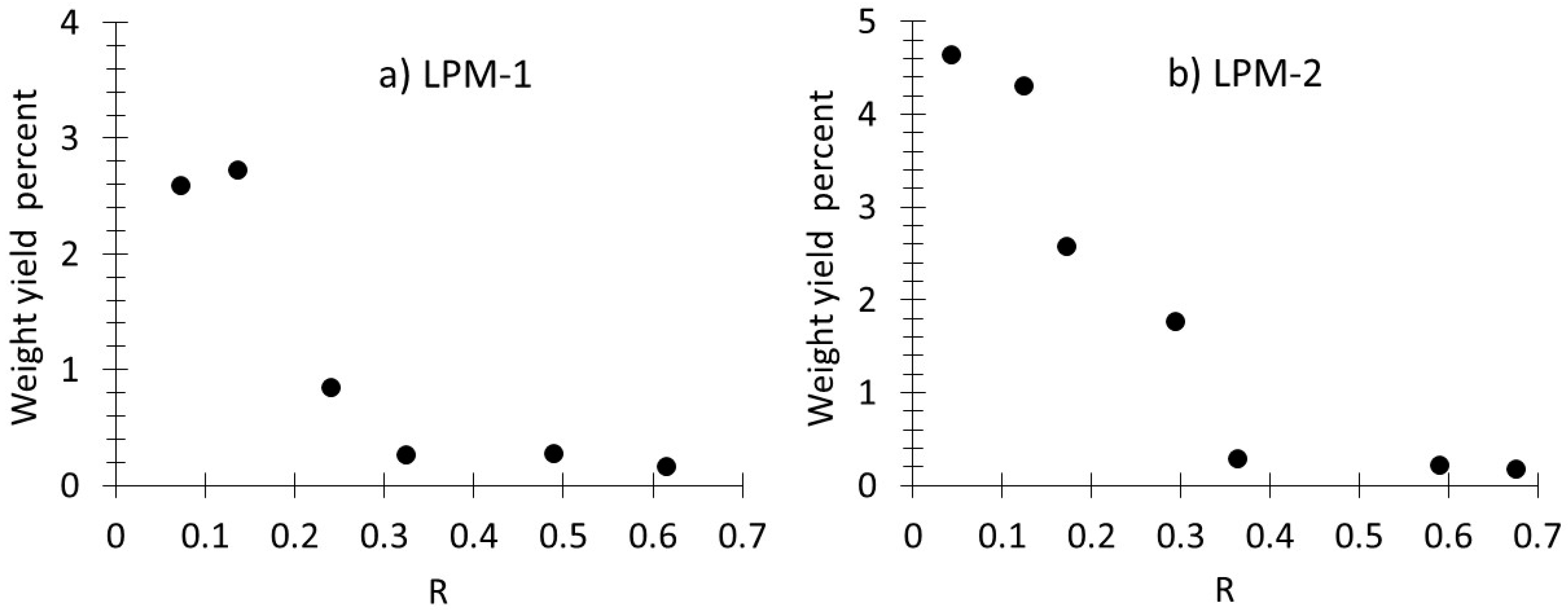
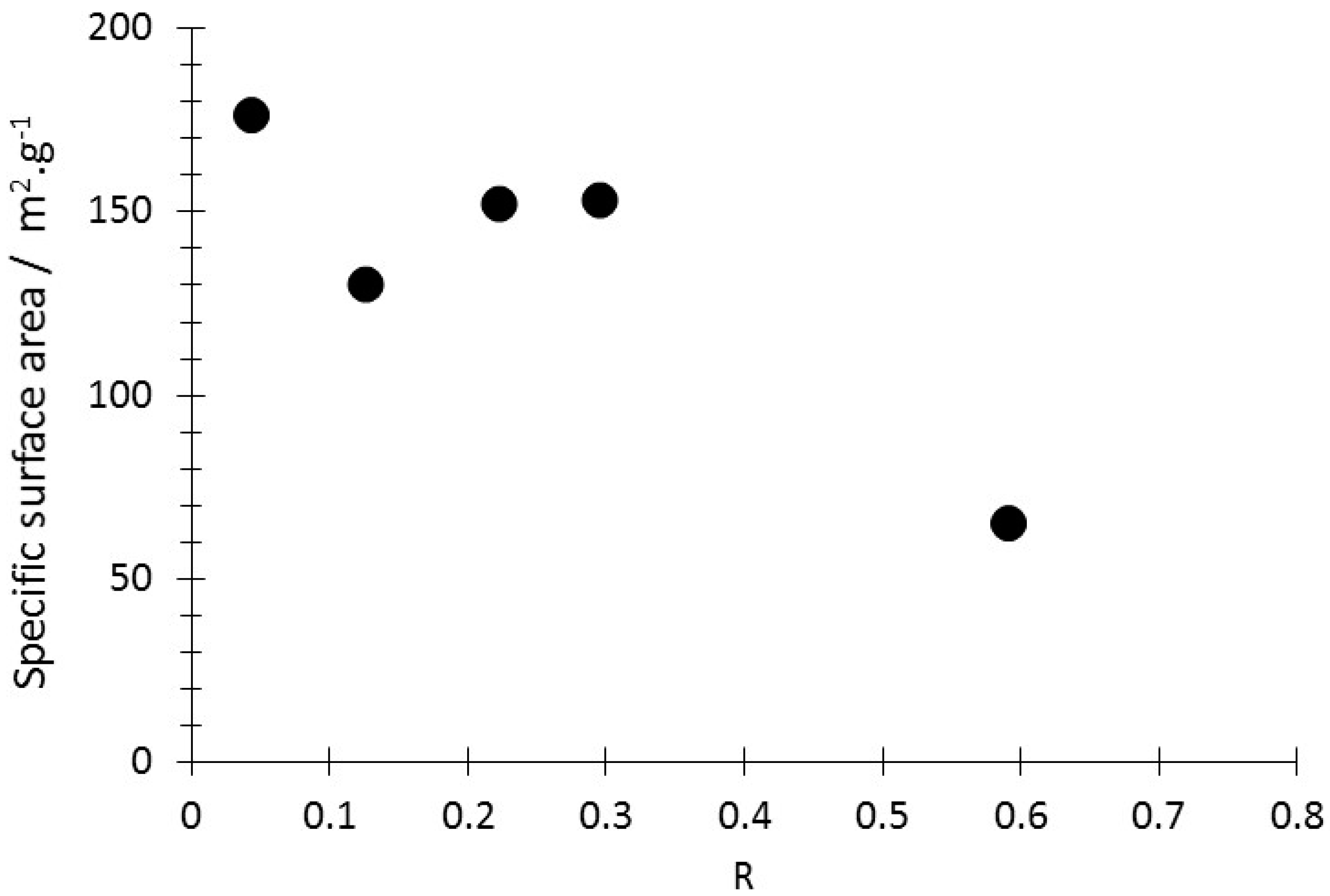
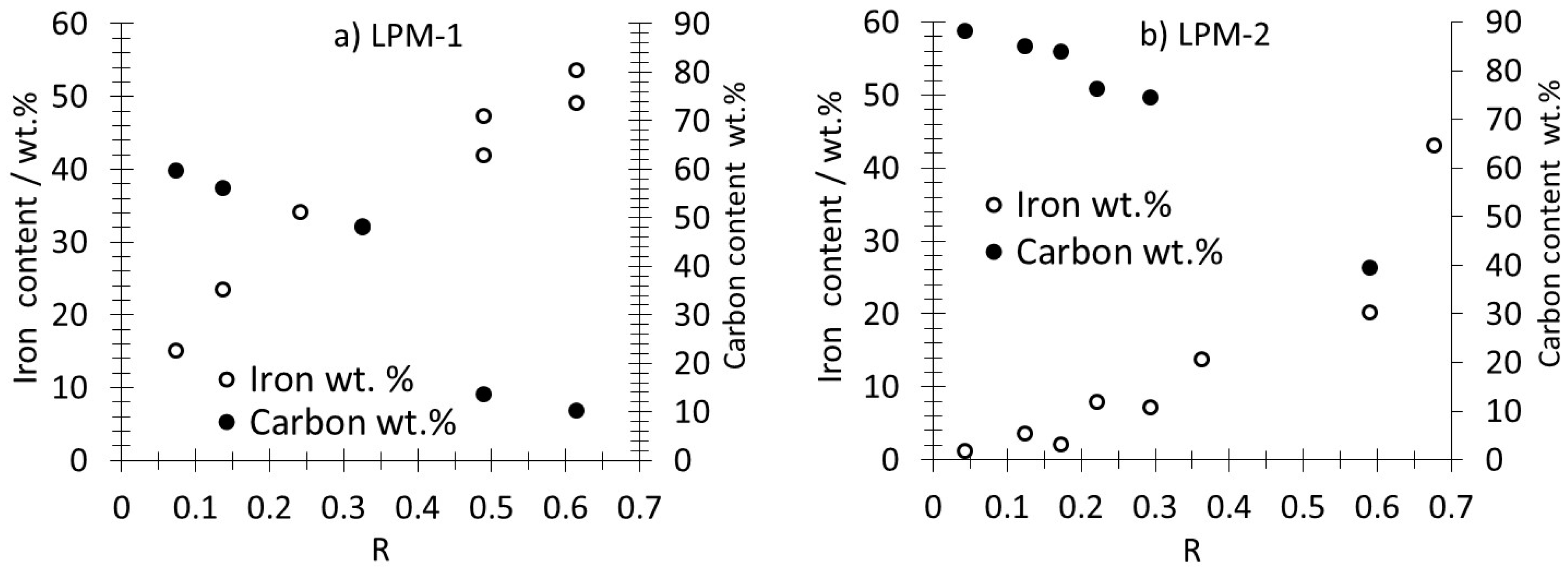
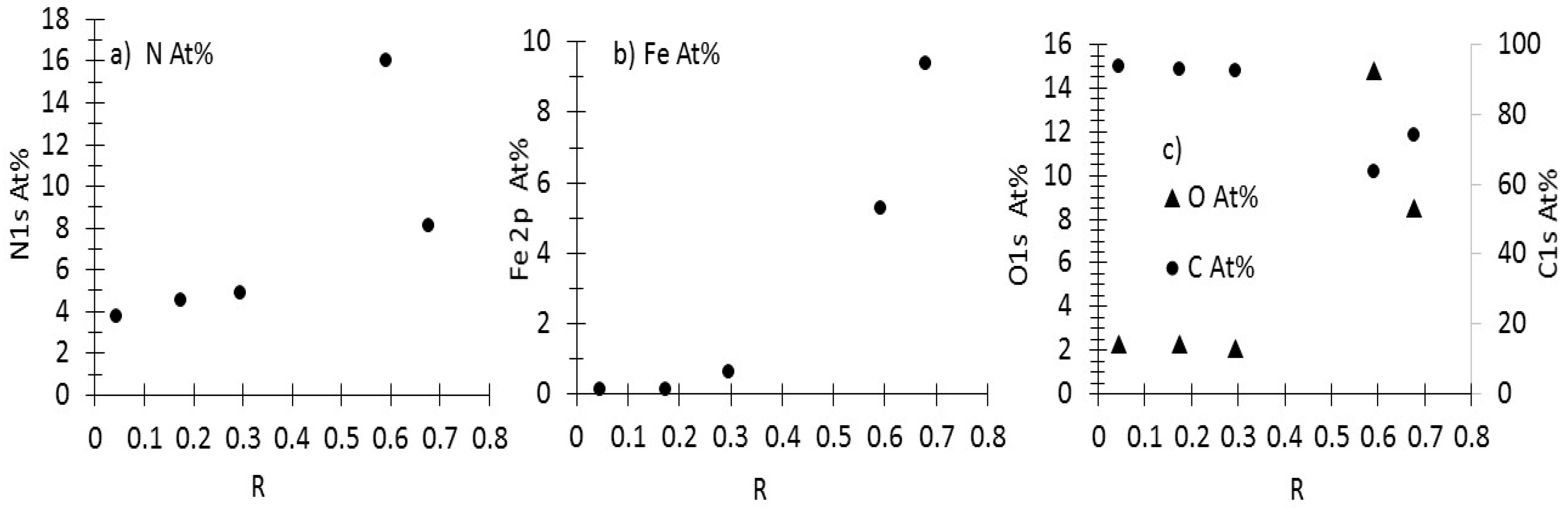
3.1. Production, Morphology and Composition Data of the Materials as a Function of R
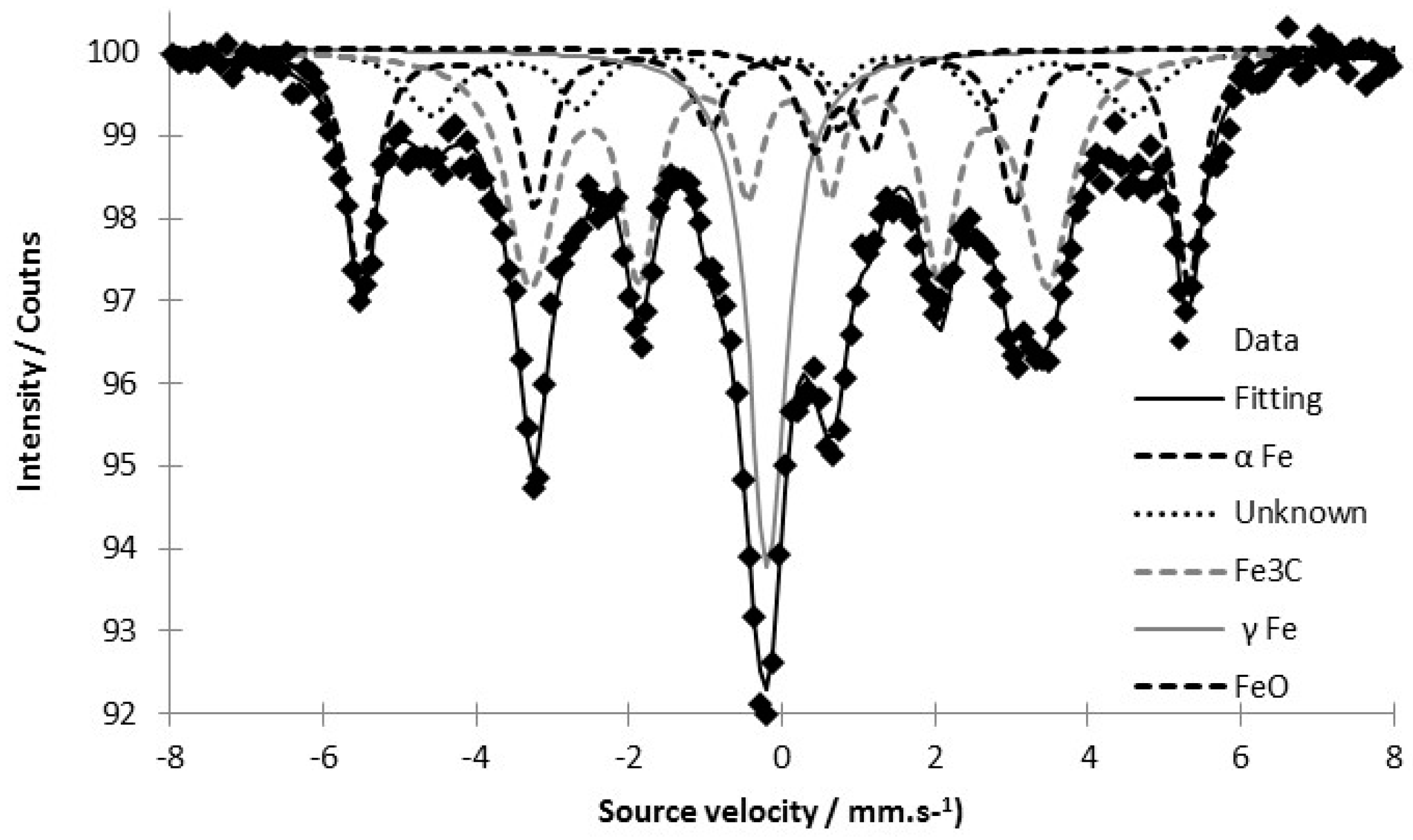
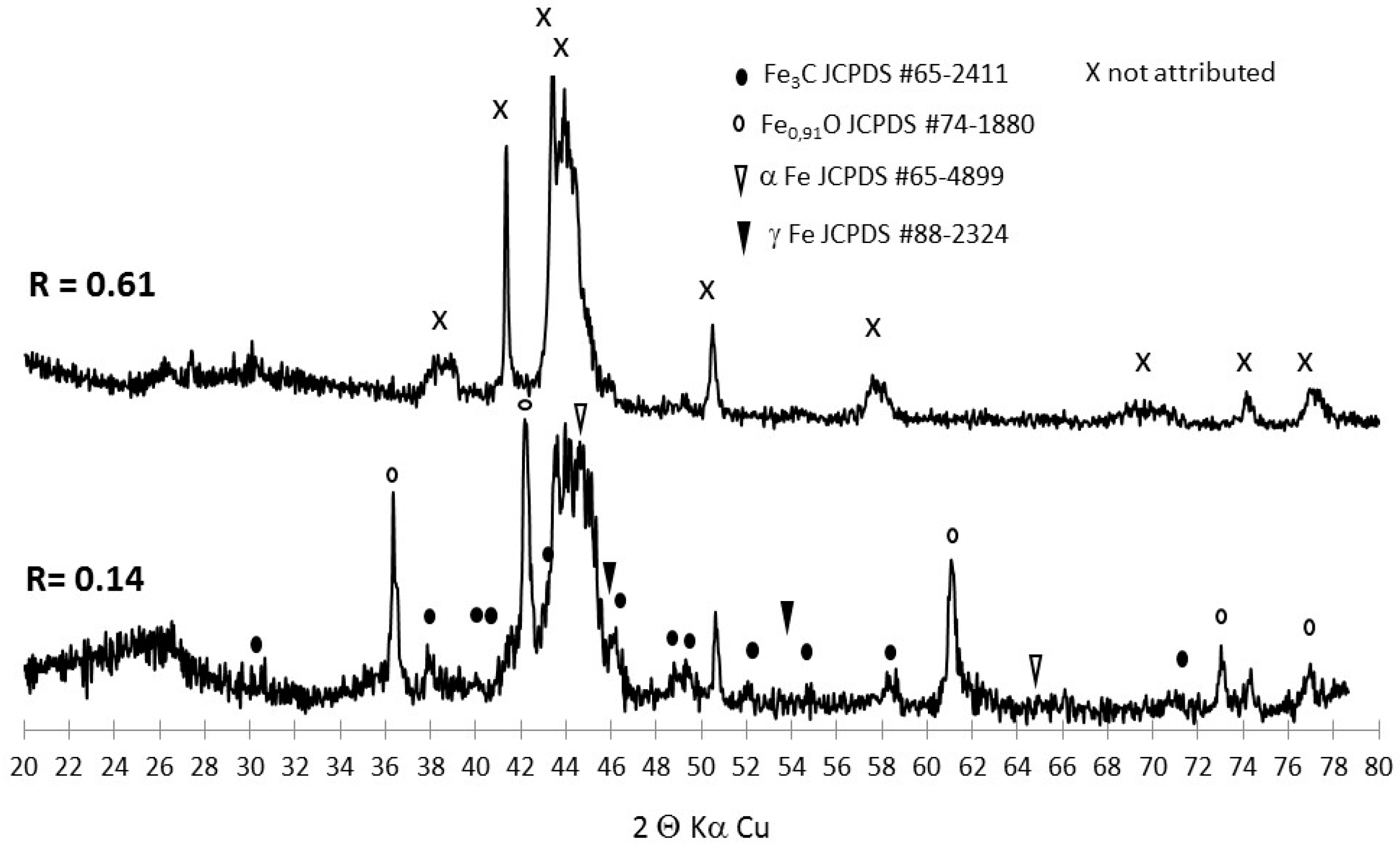
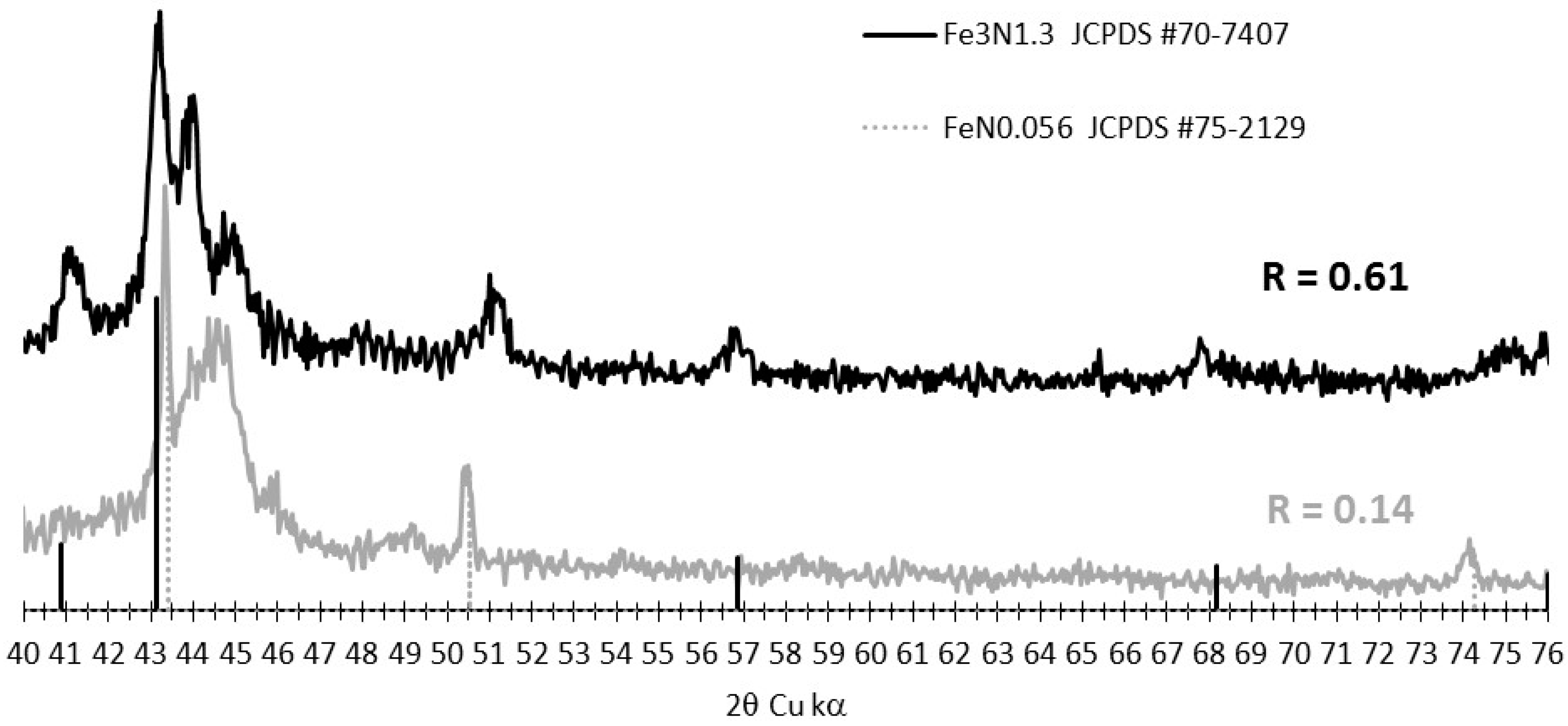
3.2. Tracking the Nature of the Iron Phases
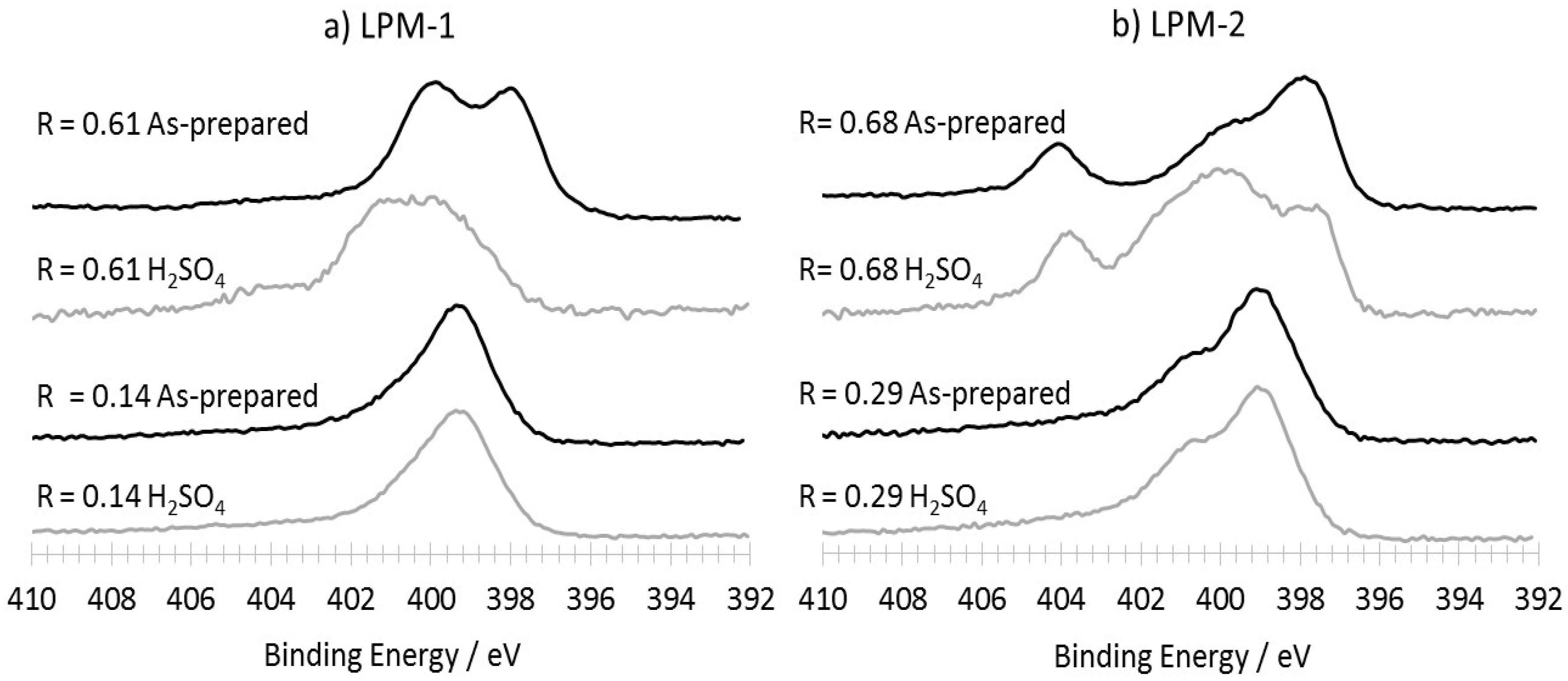
3.3. XPS Analysis
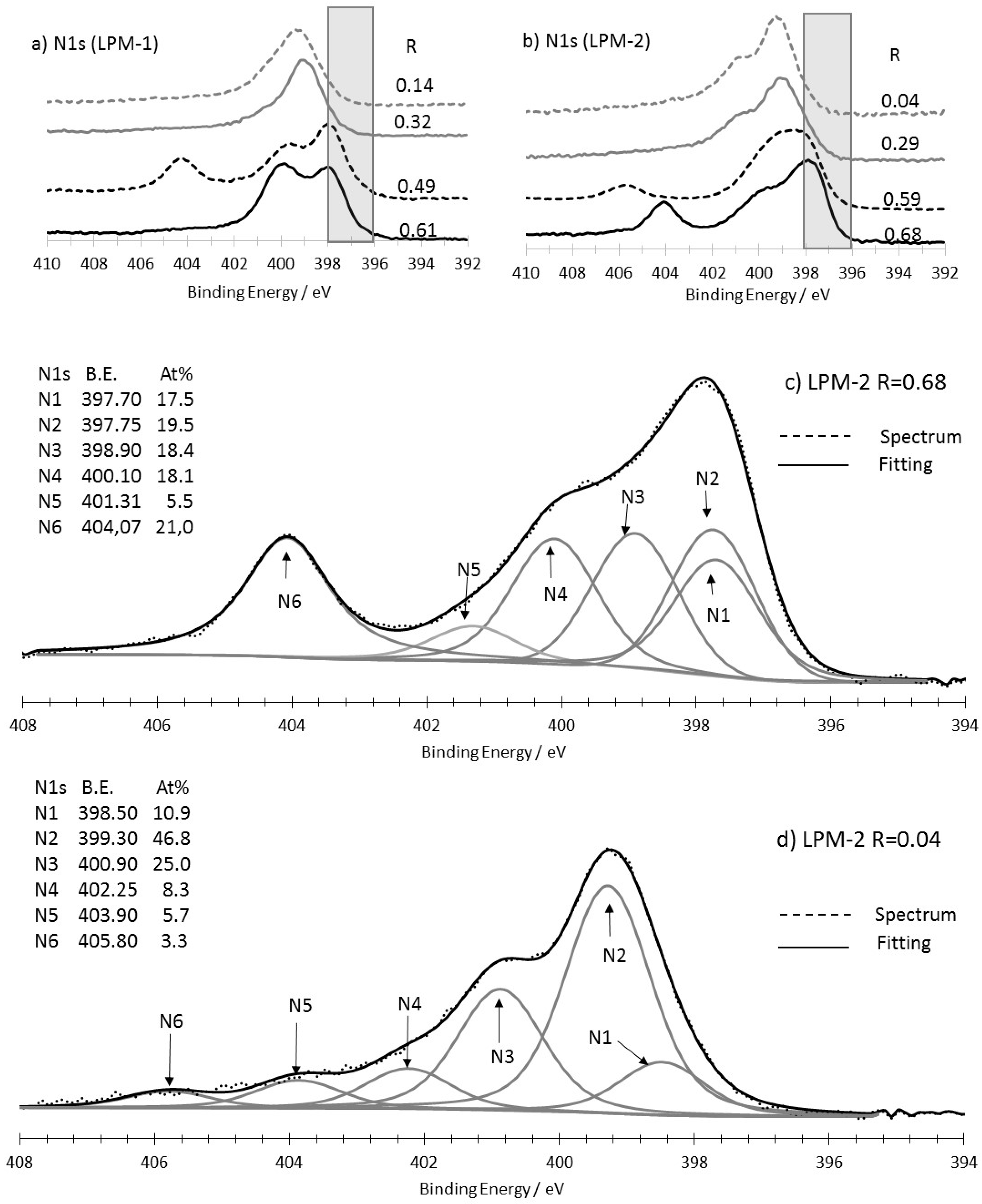
3.3.1. As-Prepared Materials
3.3.2. Characterization of the Materials Obtained from LPM-1 after Acidic Treatment
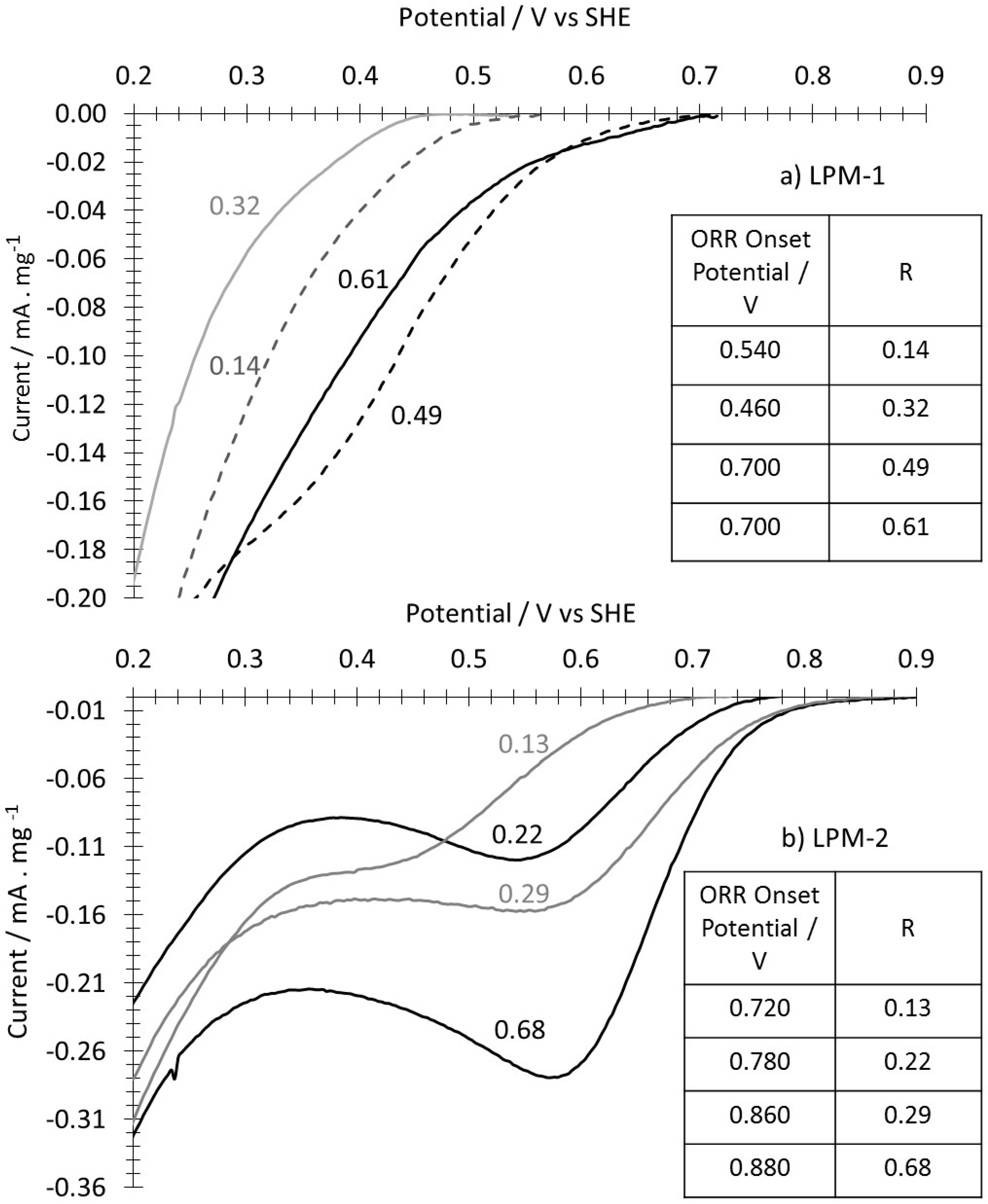
4. Preliminary Oxygen Reduction Study
5. Discussion
- -
- Heating decomposes precursors to monomers which upon saturation grow by molecular coagulation or nucleation, to critical/primary particles.
- -
- Surface reaction of monomers on primary particles can contribute to their growth, while evaporation/sublimation contribute to particle size decrease and forming monomers again.
- -
- Further coagulation and/or coalescence process between primary particles may also contribute to nanoparticles growth.
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Iron-Based Phase | Hyperfine Field—H(T) | Isomer Shift mm·s−1 | Area % |
|---|---|---|---|
| α Fe | 33.59 | 0.02 | 24.3 |
| γ Fe | - | −0.056 | 16.5 |
| Fe3C | 20.90 | 0.196 | 42.8 |
| FeO | - | 0.897 | 5.0 |
| Unknown phase | 28.33 | 0.11 | 11.4 |
Appendix B

Appendix C

References
- Jasinski, R. A New Fuel Cell Cathode Catalyst. Nature 1964, 201, 1212–1213. [Google Scholar] [CrossRef]
- Jahnke, H.; Schönborn, M.; Zimmermann, G. Organic dyestuffs as catalysts for fuel cells. Top. Curr. Chem. 1976, 61, 133–181. [Google Scholar] [PubMed]
- Gupta, S.; Tryk, D.; Bae, I.; Aldred, W.; Yeager, E. Heat-treated polyacrylonitrile-based catalysts for oxygen electroreduction. J. Appl. Electrochem. 1989, 19, 19–27. [Google Scholar] [CrossRef]
- Lefèvre, M.; Proietti, E.; Jaouen, F.; Dodelet, J.-P. Iron-Based Catalysts with Improved Oxygen Reduction Activity in Polymer Electrolyte Fuel Cells. Science 2009, 324, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Masa, J.; Xia, W.; Mulher, M.; Schuhmann, W. On the role of Metals in the Nitrogen-doped carbon electrocatalyst for oxygen reduction. Angew. Chem. Int. Ed. 2015, 54, 10102–10120. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zelenay, P. Nanostructured Nonprecious Metal Catalysts for Oxygen Reduction Reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Dombrovskis, J.K.; Palmqvist, A.E.C. Recent Progress in Synthesis, Characterization and Evaluation of Non-Precious Metal Catalysts for the Oxygen Reduction Reaction. Fuel Cells 2016, 16, 4–22. [Google Scholar] [CrossRef]
- Liu, J.; Li, E.; Ruan, M.; Song, P.; Xu, W. Recent Progress on Fe/N/C Electrocatalysts for the Oxygen Reduction Reaction in Fuel Cells. Catalysts 2015, 5, 1167–1192. [Google Scholar] [CrossRef] [Green Version]
- Russel, D.K. Infrared laser powered homogeneous pyrolysis. Chem. Soc. Rev. 1990, 19, 407–437. [Google Scholar] [CrossRef]
- David, B.; Scheneeweiss, O.; Pizurova, N.; Klementova, M.; Bezdicka, P.; Alexandrescu, R.; Dimitrache, F.; Morjan, I. Fe3C nanopowder synthesized by laser pyrolysis and its annealing behaviour. Surf. Interface Anal. 2006, 38, 482–485. [Google Scholar] [CrossRef]
- Leconte, Y.; Maskrot, H.; Combemale, L.; Herlin-Boime, N.; Reynaud, C. Application of the laser pyrolysis to the synthesis of SiC, TiC and ZrC pre-ceramics nanopowders. J. Anal. Appl. Pyrolysis 2007, 79, 465–470. [Google Scholar] [CrossRef]
- Grimes, C.A.; Qian, D.; Dickey, E.C.; Allen, J.L.; Eklund, P.C. Laser Pyrolysis fabrication of ferromagnetic γ’-Fe4N and Fe3C nanoparticles. J. Appl. Phys. 2000, 87, 5642–5644. [Google Scholar] [CrossRef] [PubMed]
- Leconte, Y.; Veintemillas-Verdaguer, S.; Morales, M.P.; Costo, R.; Rodríguez, I.; Bonville, P.; Bouchet-Fabre, B.; Herlin-Boime, N. Continuous production of water dispersible carbon–iron nanocomposites by laser pyrolysis: Application as MRI contrasts. J. Colloid Interface Sci. 2007, 313, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Pignon, B.; Miao, B.; Coste-Leconte, S.; Leconte, Y.; Marguet, S.; Jegou, P.; Bouchet-Fabre, B.; Reynaud, C.; Herlin-Boime, N. N-Doped Titanium Monoxide Nanoparticles with TiO Rock-Salt Structure, Low Energy Band Gap and Visible Light Activity. Chem. Mater. 2010, 22, 3704–3711. [Google Scholar] [CrossRef]
- Bomati-Miguel, O.; Tartaj, P.; Morales, M.P.; Bonville, P.; Golla-Schindler, U.; Zhao, X.Q.; Veintemillas-Verdaguer, S. Core–Shell Iron–Iron Oxide Nanoparticles Synthesized by Laser-Induced Pyrolysis. Small 2006, 2, 1476–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrescu, R.; Morjan, I.; Tomescu, A.; Simion, C.; Scarisoreanu, M.; Birjega, R.; Fleaca, C.; Gavrila, L.; Soare, I.; Dumitrache, F. Direct production of a novel iron-based nanocomposite from the laser pyrolysis of Fe(CO)5/MMA mixtures: Structural and sensing properties. J. Nanomater. 2010, 2010, 1–12. [Google Scholar] [CrossRef]
- Sourice, J.; Bordes, A.; Boulineau, A.; Alper, J.P.; Franger, S.; Quinsac, A.; Habert, A.; De Vito, Y.E.; Porcher, W.; Reynaud, C.; et al. Core–shell amorphous silicon–carbon nanoparticles for high performance anodes in lithium ion batteries. J. Power Sources 2016, 328, 527–535. [Google Scholar] [CrossRef]
- Wakizaka, Y.; Shishikura, T. Process for Production and Use of Carbonitride Mixture Particles or Oxycarbonitride Mixture Particles. U.S. Patent 2011/0183234, 28 July 2011. [Google Scholar]
- Cheng, X.; Than, X.-T.; Pinault, M.; Mayne, M.; Reynaud, C.; Vigneron, J.; Etcheberry, A.; Perez, H. Determination of selectivity and specific area related to oxygen reduction reaction as a function of catalyst loading on non-noble metal based electrocatalyst porous electrodes: An example on nitrogen doped carbon nanotube. Electrochim. Acta 2014, 135, 293–300. [Google Scholar] [CrossRef]
- Baret, B.; Aubert, P.-H.; Mayne-L’Hermite, M.; Pinault, M.; Reynaud, C.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes part I: Tuneable low platinum loadings, specific H upd feature and evidence for oxygen reduction. Electrochim. Acta 2009, 54, 5421–5430. [Google Scholar] [CrossRef]
- March, G.; Volatron, F.; Lachaud, F.; Cheng, X.; Baret, B.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically capped platinum nanoparticles and carbon nanotubes. Part II: Determination of diffusion area for oxygen reduction reflects platinum accessibility. Electrochim. Acta 2011, 56, 5151–5157. [Google Scholar] [CrossRef]
- Cheng, X.; Volatron, F.; Pardieu, E.; Borta, A.; Carrot, G.; Reynaud, C.; Mayne, M.; Pinault, M.; Etcheberry, A.; Perez, H. Nanocomposite electrodes based on pre-synthesized organically grafted platinum nanoparticles and carbon nanotubes III. Determination of oxygen reduction reaction selectivity and specific area of porous electrode related to the oxygen reduction reaction ranging from 2 m2·gPt−1 to 310 m2·gPt−1. Electrochim. Acta 2013, 89, 1–12. [Google Scholar] [CrossRef]
- Oya, A.; Marsh, H. Phenomena of catalytic graphitization. J. Mater. Sci. 1982, 17, 309–322. [Google Scholar] [CrossRef]
- Hargreaives, J.S.J. Heterogeneous catalysis with metal nitrides. Coord. Chem. Rev. 2013, 257, 2015–2031. [Google Scholar] [CrossRef]
- Han, Y.; Wang, H.; Zang, M.; Su, M.; Li, W.; Tao, K. Low-temperature approach to synthesize iron nitride from amorphous Iron. Inorg. Chem. 2008, 47, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, W.; Panzner, G.; Grabke, H.J. The bonding state of nitrogen segregated on Fe (100) and on iron nitrides Fe4N and Fe2N. Surf. Sci. 1989, 218, 507–518. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hong, J.-Y.; Yen, C.-H.; Tong, S.-Y.; Tung, M.-J.; Shiu, H.-W.; Lin, M.-T. X-ray photelectron spectroscopic investigation on Fe geometrical sites of iron nitride thin films. Jpn. J. Appl. Phys. 2015, 54, 033002. [Google Scholar] [CrossRef]
- Torres, J.; Perry, C.C.; Bransfield, S.J.; Fairbrother, D.H. Low temperature oxidation of nitride iron surfaces. J. Phys. Chem. B 2003, 107, 5558–5567. [Google Scholar] [CrossRef]
- Susi, T.; Pichler, T.; Ayala, P. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. Beilstein J. Nanotechnol. 2015, 6, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Casolo, S.; Suzuki, T.; Shikano, T.; Sakurai, M.; Harada, Y.; Saito, M.; Oshima, M.; Trioni, M.I.; Tantardini, G.F.; et al. Atomic-scale characterization of nitrogen-doped graphite: Effects of dopant nitrogen on the local electronic structure of the surrounding carbon atoms. Phys. Rev. B 2012, 86, 035436. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Kapteijn, F.; Moulijn, J.A.; Matzner, S.; Boehm, H.-P. The development of nitrogen functionality in model chars during gasification in CO2 and O2. Carbon 1999, 37, 1143–1150. [Google Scholar] [CrossRef]
- Marton, D.; Boyd, K.J.; Al-Bayati, A.H.; Todorov, S.S.; Rabalais, J.W. Carbon Nitride Deposited Using Energetic Species: A Two-Phase System. Phys. Rev. Lett. 1994, 73, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.C.; Park, J. Distribution and Structure of N Atoms in Multiwalled Carbon Nanotubes Using Variable-Energy X-ray Photoelectron Spectroscopy. J. Phys. Chem. B 2005, 109, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Gammon, W.J.; Kraft, O.; Reilly, A.C.; Holloway, B.C. Experimental comparison of N(1s) X-ray photoelectron spectroscopy binding energies of hard and elastic amorphous carbon nitride films with reference organic compounds. Carbon 2003, 41, 1917–1923. [Google Scholar] [CrossRef]
- Artyushkova, K.; Serov, A.; Rojas-Carbonell, S.; Atanassov, P. Chemistry of Multitudinous Active Sites for Oxygen Reduction Reaction in Transition Metal−Nitrogen−Carbon. J. Phys. Chem. C 2015, 119, 25917–25928. [Google Scholar] [CrossRef]
- Casanovas, J.; Ricart, J.M.; Rubio, J.; Illas, F.; Jimenez-Mateos, J.M. Origin of the Large N1s Binding Energy in X-ray Photoelectron Spectra of Calcined Carbonaceous Materials. J. Am. Chem. Soc. 1996, 118, 8071–8076. [Google Scholar] [CrossRef]
- Jimenez Mateos, J.M.; Fierro, J.L.G. X-ray Photoelectron Spectroscopic Study of Petroleum Fuel Cokes. Surf. Int. Anal. 1996, 2, 223–236. [Google Scholar] [CrossRef]
- Buesser, B.; Pratsinis, S.E. Design of Nanomaterial Synthesis by Aerosol Processes. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 103–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buesser, B.; Gröhn, A.J. Multiscale aspect of modeling gas phase nanoparticle synthesis. Chem. Eng. Technol. 2012, 35, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, G.A.; Goudeli, E.; Pratsinis, S.E. Flame synthesis of functional nanostructured materials and devices: Surface growth and aggregation. Proc. Energy Combust. Sci. 2017, 36, 29–50. [Google Scholar] [CrossRef]
- Strobel, R.; Pratsinis, S.E. Flame aerosol synthesis of smart nanostructured materials. J. Mater. Chem. 2007, 17, 4743–4756. [Google Scholar] [CrossRef]
- Teoh, W.Y.; Amal, R.; Mädler, L. Flame spray pyrolysis: An enabling technology for nanoparticles design and fabrication. Nanoscale 2010, 2, 1324–1347. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, A.; Megaridis, C.M. Morphology of Flame-Generated Soot As Determined by Thermophoretic Sampling. Langmuir 1987, 3, 254–259. [Google Scholar] [CrossRef]
- Castro, C. Mécanismes de Croissance de Nanotubes de Carbone Alignés: Relation Catalyseur—Nanotube. Ph.D. Thesis, Paris XI Orsay University, Orsay, France, 2009. [Google Scholar]
- Jaouen, F.; Proietti, E.; Lefèvre, M.; Chentiz, R.; Dodelet, J.-P.; Wu, G.; Chung, H.T.; Johnston, P.; Zelenay, C.M. Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ. Sci. 2011, 4, 114–130. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Tylus, U.; Strickland, K.; Li, J.; Serov, A.; Artyushkova, K.; Atanassov, P.; Anibal, J.; Gumeci, C.; et al. Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid. Nano Energy 2016, 29, 65–82. [Google Scholar] [CrossRef]
- Jia, Q.; Ramaswamy, N.; Hafiz, H.; Tylus, U.; Strickland, K.; Wu, G.; Barbiellini, B.; Bansil, A.; Holby, E.F.; Zelenay, P.; et al. Experimental Observation of Redox-Induced Fe–N Switching Behavior as a Determinant Role for Oxygen Reduction Activity. ACS Nano 2015, 9, 12496–12505. [Google Scholar] [CrossRef] [PubMed]
- Daems, N.; Sheng, X.; Vankelecom, I.F.J.; Pescarmona, P. Metal-free doped carbon materials as electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2014, 2, 4085–4110. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jensen, J.O.; Zhang, W.; Cleemann, L.N.; Xing, W.; Bjerrum, N.J.; Li, Q. Hollow Spheres of Iron Carbide Nanoparticles Encased in Graphitic Layers as Oxygen Reduction Catalysts. Angew. Chem. Int. Ed. 2014, 53, 3675–3679. [Google Scholar] [CrossRef] [PubMed]
- Kramm, U.I.; Herrman-Geppert, I.; Bogdanoff, P.; Fiechter, S. Effect of an ammonia treatment on structure, composition and oxygen reduction reaction activity of Fe–N–C catalysts. J. Phys. Chem. C 2011, 115, 23417. [Google Scholar] [CrossRef]
- Ron, M.; Mathalone, Z. Hyperfine Interactions of 57Fe in Fe3C. Phys. Rev. B 1971, 4, 774–777. [Google Scholar] [CrossRef]
- Pasternak, M.P.; Taylor, R.D.; Jeanloz, R.; Li, X.; Nguyen, J.H.; McCammon, C.A. High Pressure Collapse of Magnetism in Fe0.94O: Mössbauer Spectroscopy Beyond 100 GPa. Phys. Rev. Lett. 1997, 79, 5046–5049. [Google Scholar] [CrossRef]
- Serov, A.; Artyushkova, K.; Niangar, E.; Wang, C.; Dale, N.; Jaouen, F.; Sougrati, M.-T.; Jia, Q.; Mukerjee, S.; Atanassov, P. Nano-Structured Non-Platinum Catalysts for Automotive Fuel Cell Application. Nano Energy 2015, 16, 293–300. [Google Scholar] [CrossRef]












| LPM-1 | LPM-1 | LPM-2 | LPM-2 | |||||
|---|---|---|---|---|---|---|---|---|
| R = 0.61 | R = 0.14 | R = 0.68 | R = 0.29 | |||||
| N at % | Fe at % | N at % | Fe at % | N at % | Fe at % | N at % | Fe at % | |
| As-prepared materials | 6.7 | 3.7 | 4.6 | 0.05 | 8.1 | 9.4 | 4.9 | 0.6 |
| H2SO4 treated | 2.0 | 0.3 | 4.2 | 0 | 6.0 | 3.0 | 4.2 | 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, H.; Jorda, V.; Bonville, P.; Vigneron, J.; Frégnaux, M.; Etcheberry, A.; Quinsac, A.; Habert, A.; Leconte, Y. Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C 2018, 4, 43. https://doi.org/10.3390/c4030043
Perez H, Jorda V, Bonville P, Vigneron J, Frégnaux M, Etcheberry A, Quinsac A, Habert A, Leconte Y. Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C. 2018; 4(3):43. https://doi.org/10.3390/c4030043
Chicago/Turabian StylePerez, Henri, Virginie Jorda, Pierre Bonville, Jackie Vigneron, Mathieu Frégnaux, Arnaud Etcheberry, Axelle Quinsac, Aurélie Habert, and Yann Leconte. 2018. "Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction" C 4, no. 3: 43. https://doi.org/10.3390/c4030043
APA StylePerez, H., Jorda, V., Bonville, P., Vigneron, J., Frégnaux, M., Etcheberry, A., Quinsac, A., Habert, A., & Leconte, Y. (2018). Synthesis and Characterization of Carbon/Nitrogen/Iron Based Nanoparticles by Laser Pyrolysis as Non-Noble Metal Electrocatalysts for Oxygen Reduction. C, 4(3), 43. https://doi.org/10.3390/c4030043