Design of Porous Carbons for Supercapacitor Applications for Different Organic Solvent-Electrolytes


Abstract
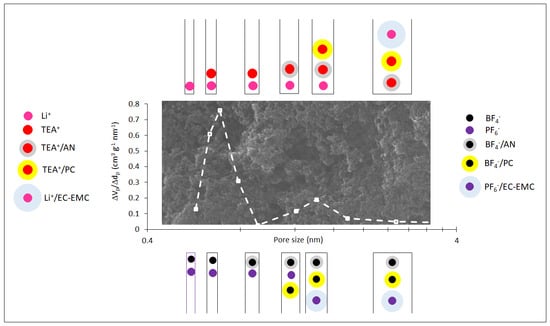
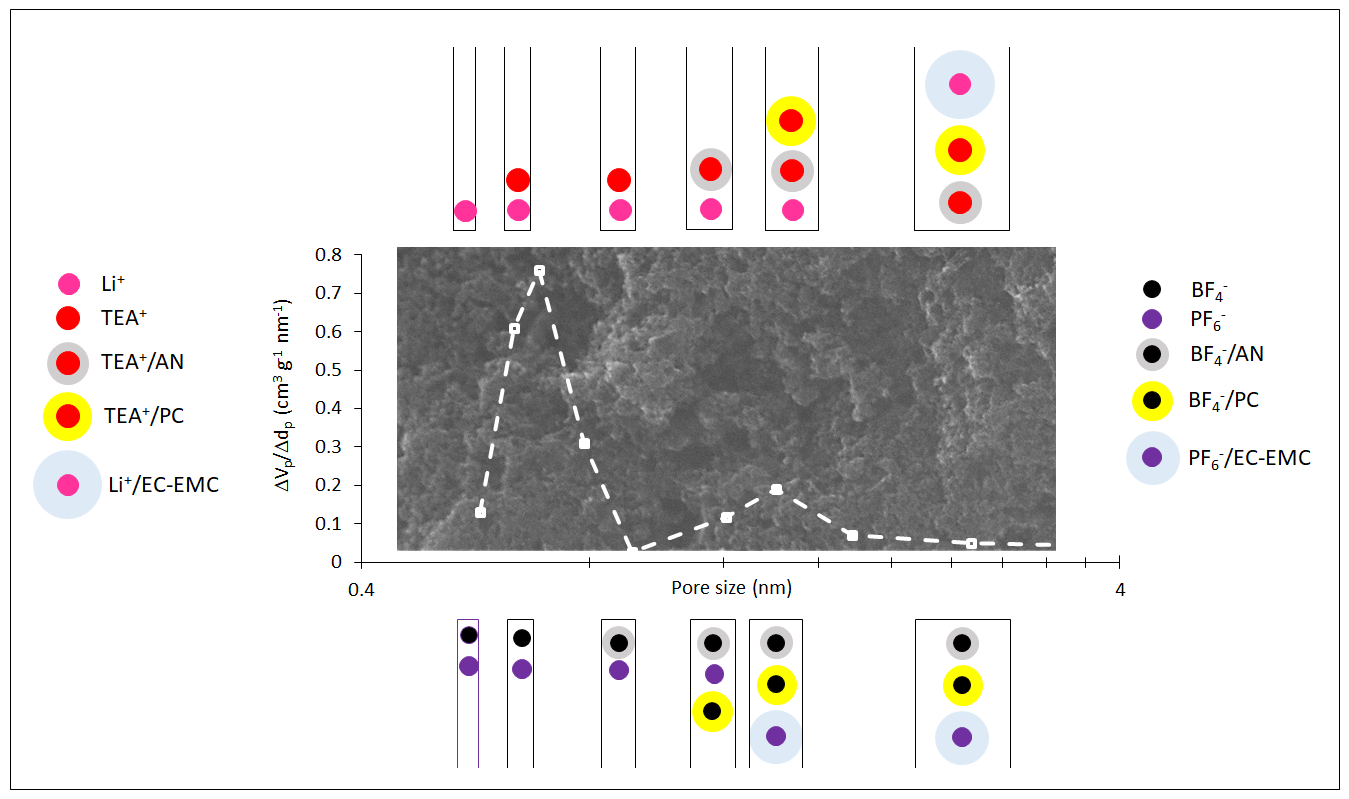
:
1. Introduction
2. Materials and Methods
- (a)
- ACF fabric ACC-507-15 (Kynol Europa Gmbh, Hamburg, Germany) of a nominal thickness of 0.5 mm and areal density of 12.05 mg cm−2;
- (b)
- AC coating electrodes consisting of 95 wt% activated carbon (AC) (Sigma Aldrich: product C9157: 4C Norit A charcoal, peat bog-derived, BETAC = 1000 m2 g−1) and 5 wt% polyvinylidene difluoride (PVDF) (MW = 534 × 103, from Sigma Aldrich, Gillingham, UK).
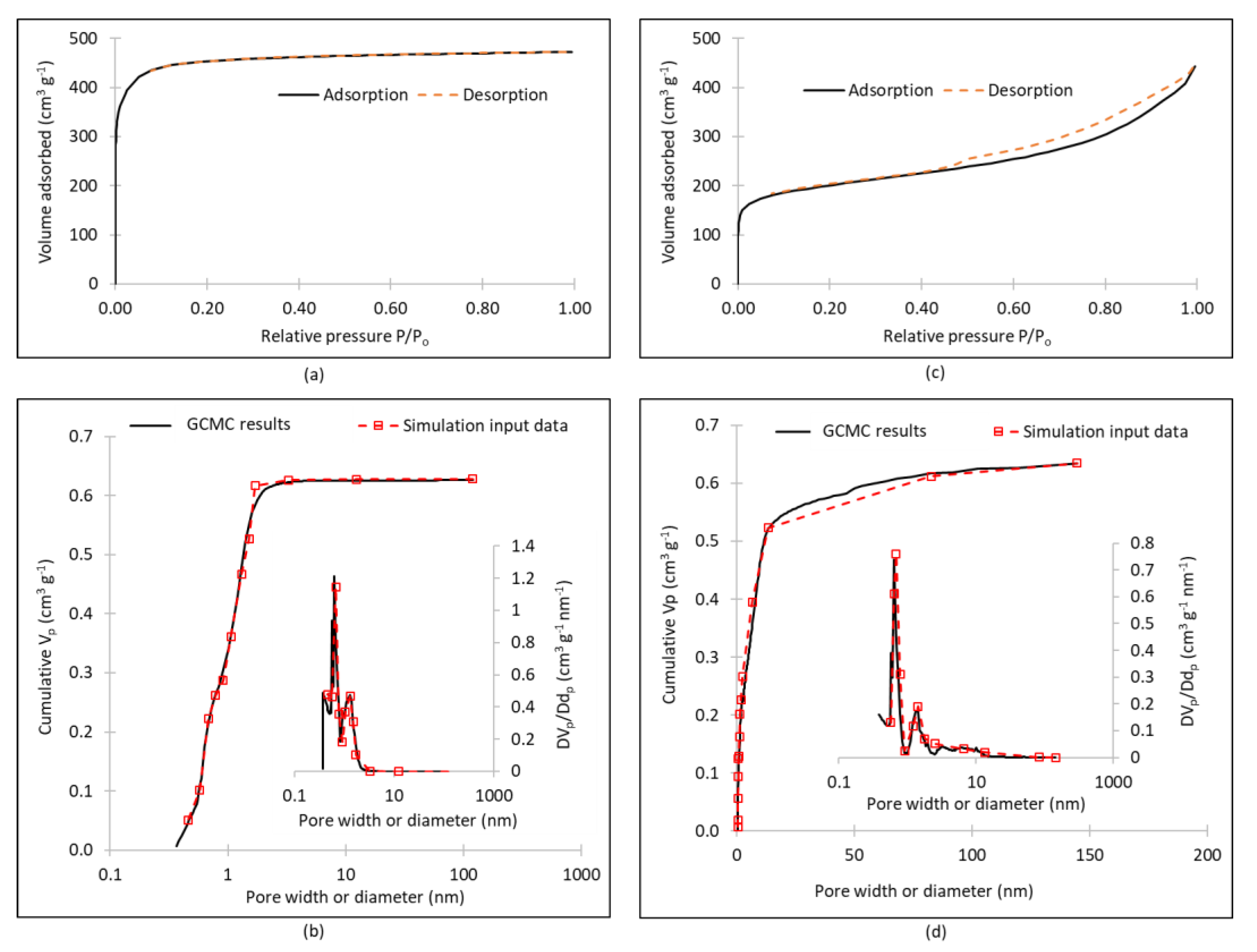
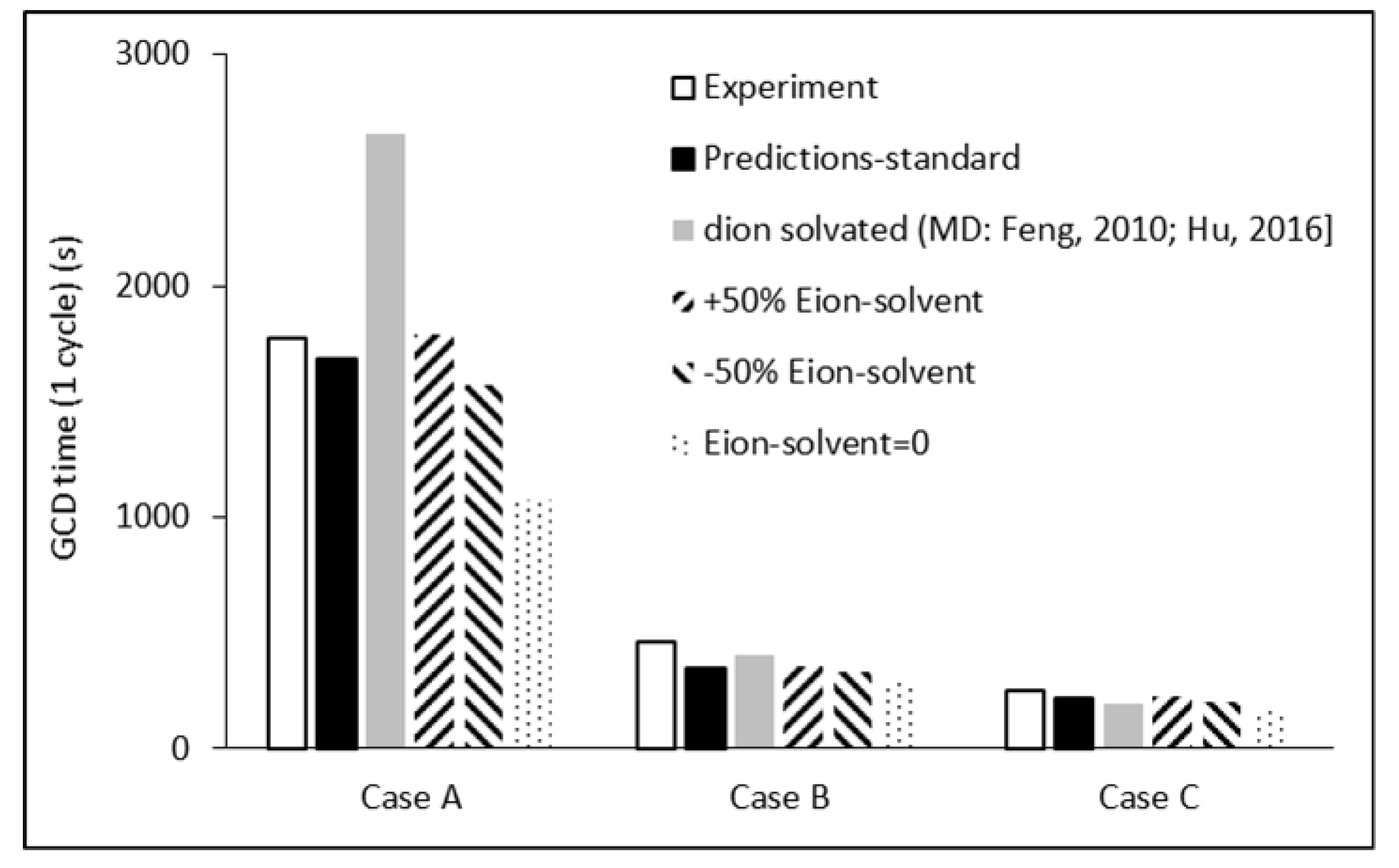
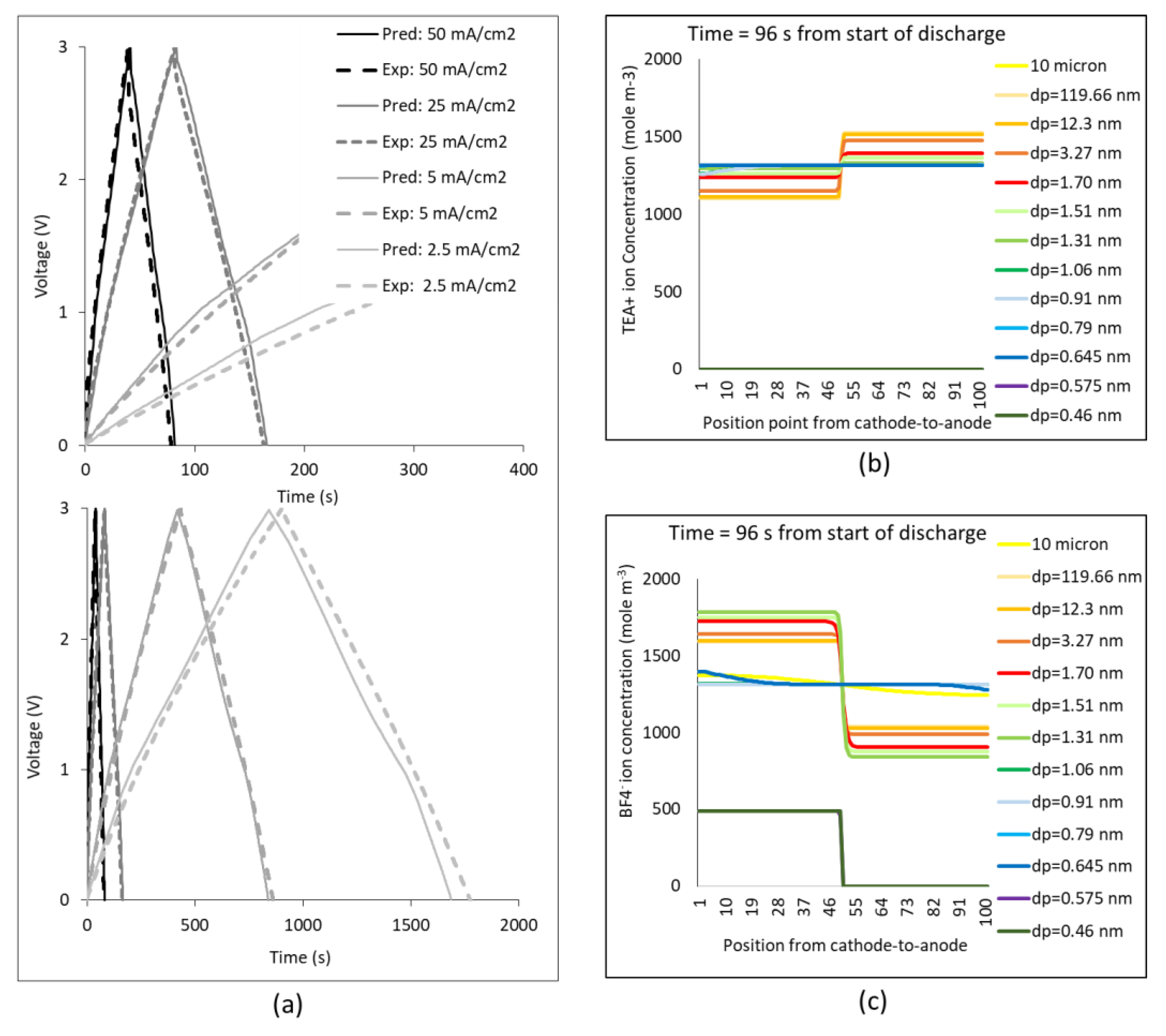
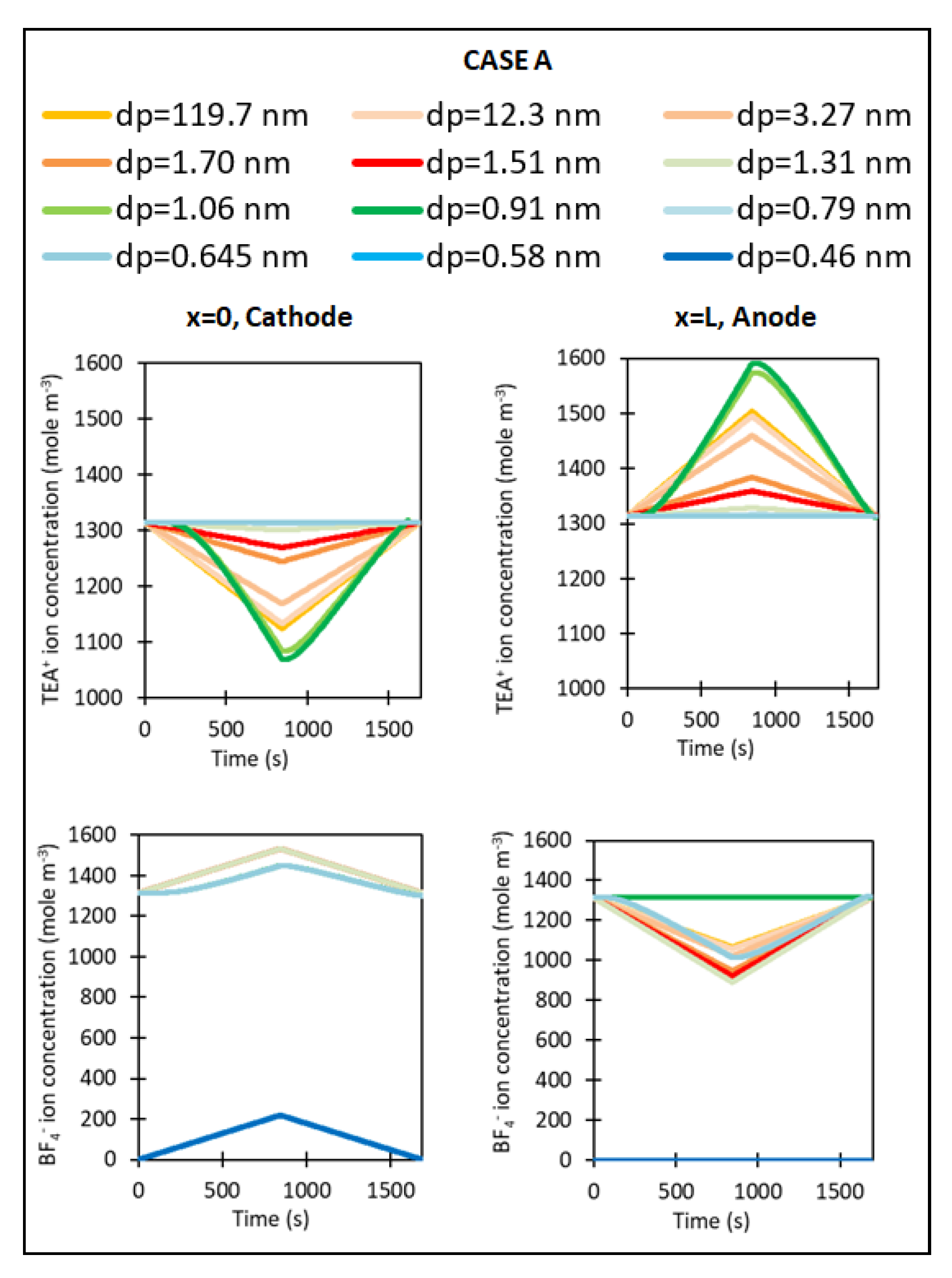
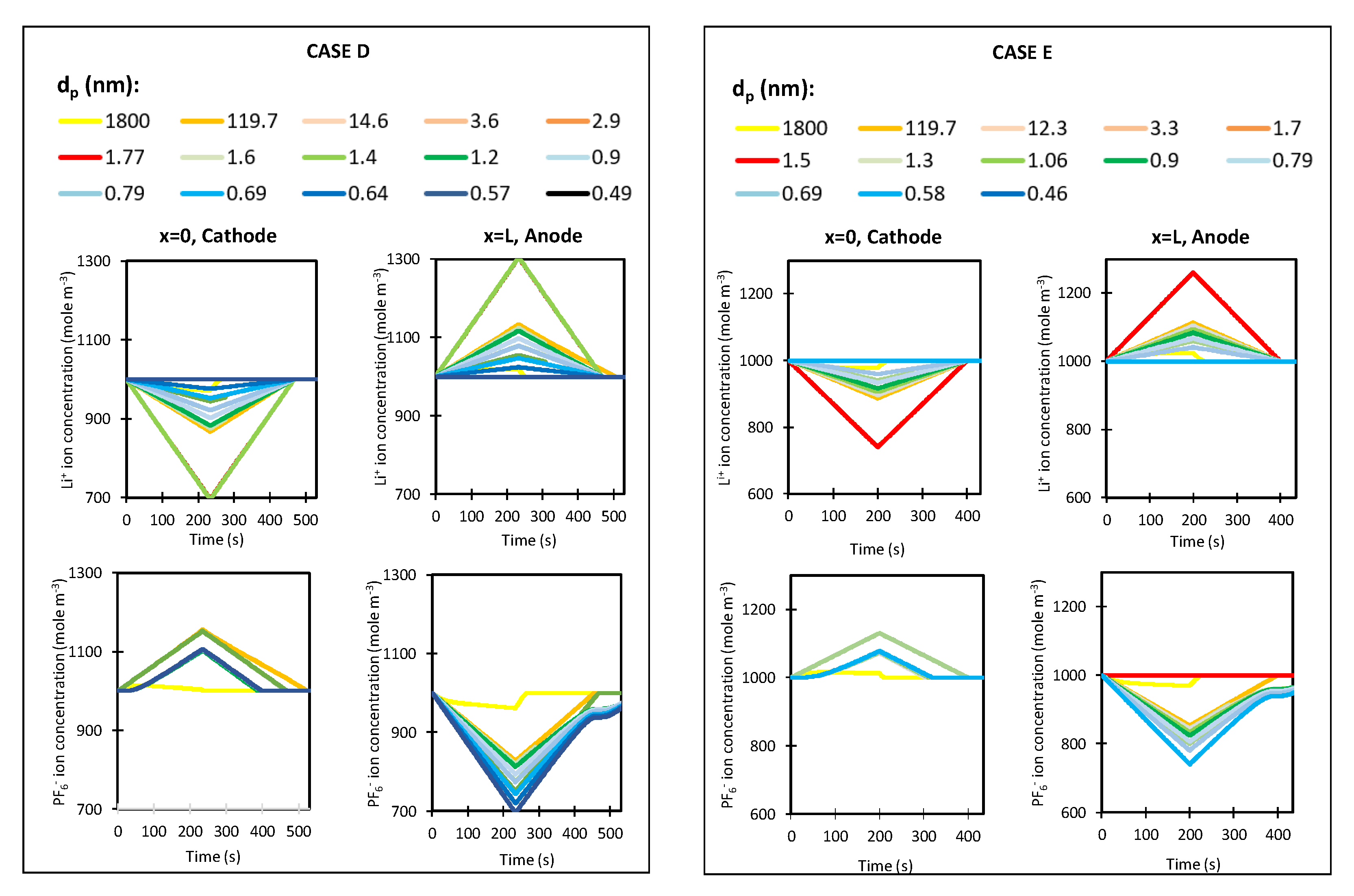
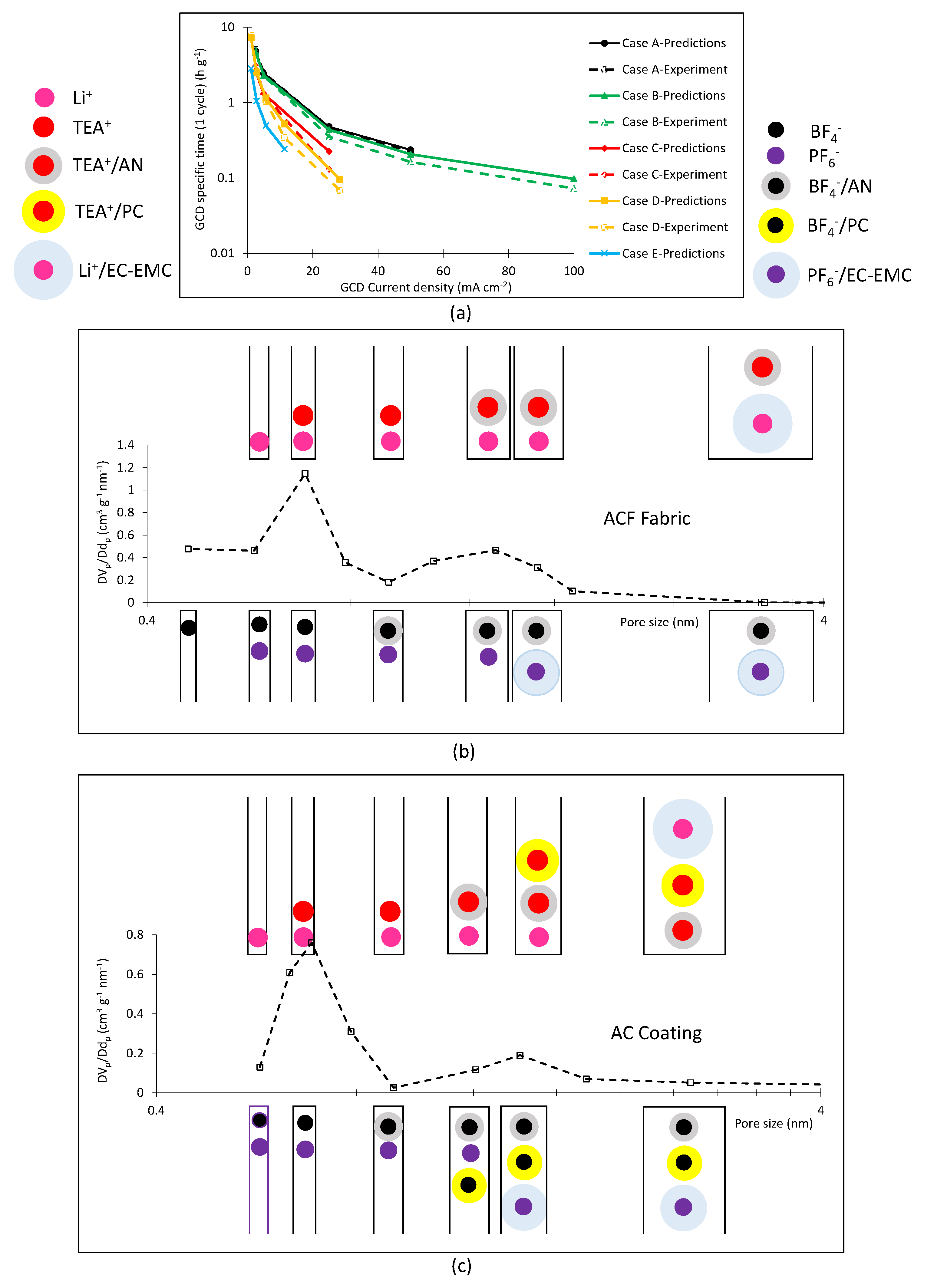
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass ion transport equations for all pore sizes p (p = 1 to Np) and ions i (i = + and −): | (A1) |
| Current flux of ion i from pore p − 1 to pore p: | (A2) |
| Decay factor if electrochemical energy, EEC, is less than : | (A3) |
| Desolvation energy barrier for dion < dp < dsolv.ion+ (for larger pores: Δni = 0): | (A4) |
| (A5) | |
| (A6) | |
| (A7) | |
| (A8) | |
| (A9) | |
| (A10) | |
| (A11) | |
| Poisson equation: | (A12) |
| (A13) | |
| (A14) | |
| (A15) | |
| (A16) | |
| (A17) | |
| (Nernst-Einstein equation) | (A18) |
| Initial conditions, at t = 0, before the first charge: for dp > dion,i and for dp < dion,i i1 = i2 = 0. ϕ1 = ϕ2 = 0. | (A19) |
| Boundary conditions: At x = xS (separator): i1 = 0 ϕ1 = 0 At x = 0 and x = L (current collector/cathode or anode): │i1│= io, the current density in galvanostatic charge-discharge. | (A20) |
References
- Simon, P.; Gogotsi, Y. Perspectives for electrochemical capacitors and related devices. Nat. Mater. 2020, 19, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Markoulidis, F.; Lei, C.; Lekakou, C. Investigations of activated carbon fabric-based supercapacitors with different interlayers via experiments and modelling of electrochemical processes of different timescales. Electrochim. Acta 2017, 249, 122–134. [Google Scholar] [CrossRef]
- Pina, A.C.; Amaya, A.; Marcuzzo, J.S.; Rodrigues, A.C.; Baldan, M.R.; Tancredi, N.; Cuña, A. Supercapacitor Electrode Based on Activated Carbon Wool Felt. C J. Carbon Res. 2018, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.; Amini, N.; Markoulidis, F.; Wilson, P.; Tennison, S.; Lekakou, C. Activated carbon from phenolic resin with controlled mesoporosity for an electric double-layer capacitor (EDLC). J. Mater. Chem. A 2013, 1, 6037–6042. [Google Scholar] [CrossRef]
- Lei, C.; Lekakou, C. Activated carbon–carbon nanotube nanocomposite coatings for supercapacitor applications. Surf. Coat. Technol. 2013, 232, 326–330. [Google Scholar] [CrossRef]
- Reece, R.; Lekakou, C.; Smith, P.A. A High-Performance Structural Supercapacitor. ACS Appl. Mater. Interfaces 2020, 12, 25683–25692. [Google Scholar] [CrossRef]
- Vermisoglou, E.C.; Giannakopoulou, T.; Romanos, G.; Giannouri, M.; Boukos, N.; Lei, C.; Lekakou, C.; Trapalis, C. Effect of hydrothermal reaction time and alkaline conditions on the electrochemical properties of reduced graphene oxide. Appl. Surf. Sci. Part A 2015, 358, 100–109. [Google Scholar] [CrossRef]
- Liu, G.; Yu, Z.; Neff, D.; Zhamu, A.; Jang, B.Z. Graphene-based supercapacitor with an ultrahigh energy density. Nano Lett. 2010, 10, 4863–4868. [Google Scholar] [CrossRef]
- Yang, H.; Kannappan, S.; Pandian, A.S.; Jang, J.-H.; Lee, Y.S.; Lu, W. Graphene supercapacitor with both high power and energy density. Nanotechnology 2017, 28, 445401. [Google Scholar] [CrossRef] [PubMed]
- Vermisoglou, E.C.; Giannakopoulou, T.; Romanos, G.; Boukos, N.; Psycharis, V.; Lei, C.; Lekakou, C.; Petridis, D.; Trapalis, C. Graphene-based materials via benzidine-assisted exfoliation and reduction of graphite oxide and their electrochemical properties. Appl. Surf. Sci. 2017, 392, 244–255. [Google Scholar] [CrossRef]
- Reece, R.; Lekakou, C.; Smith, P.A.; Grilli, R.; Trapalis, C. Sulphur-linked graphitic and graphene oxide platelet-based electrodes for electrochemical double layer capacitors. J. Alloy. Compd. 2019, 792, 582–593. [Google Scholar] [CrossRef]
- Vermisoglou, E.C.; Giannakopoulou, T.; Romanos, G.E.; Boukos, N.; Giannouri, M.; Lei, C.; Lekakou, C.; Trapalis, C. Non-activated high surface area expanded graphite oxide for supercapacitors. Appl. Surf. Sci. Part A 2015, 358, 110–121. [Google Scholar] [CrossRef]
- Lekakou, C.; Moudam, O.; Markoulidis, F.; Andrews, T.; Watts, J.F.; Reed, G.T. Carbon-based fibrous EDLC capacitors and supercapacitors. J. Nanotechnol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Izadi-Najafabadi, A.; Yamada, T.; Futaba, D.N.; Yudasaka, M.; Takagi, H.; Hatori, H.; Iijima, S.; Hata, K. High-power supercapacitor electrodes from single-walled carbon nanohorn/nanotube composite. ACS Nano 2011, 5, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Markoulidis, F.; Lei, C.; Lekakou, C. Fabrication of high-performance supercapacitors based on transversely oriented carbon nanotubes. Appl. Phys. A 2013, 111, 227–236. [Google Scholar] [CrossRef]
- Markoulidis, F.; Lei, C.; Lekakou, C.; Duff, D.; Khalil, S.; Martorana, B.; Cannavaro, I. A method to increase the energy density of supercapacitor cells by the addition of multiwall carbon nanotubes into activated carbon electrodes. Carbon 2014, 68, 58–66. [Google Scholar] [CrossRef]
- Markoulidis, F.; Todorova, N.; Grilli, R.; Lekakou, C.; Trapalis, C. Composite electrodes of activated carbon and multiwall carbon nanotubes decorated with silver nanoparticles for high power energy storage. J. Compos. Sci. 2019, 3, 97. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.; Lei, C.; Markoulidis, F.; Lekakou, C. The Composite supercapacitor. Energy Technol. 2016, 4, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Vermisoglou, E.C.; Giannouri, M.; Todorova, N.; Giannakopoulou, T.; Lekakou, C.; Trapalis, C. Recycling of typical supercapacitor materials. Waste Manag. Res. 2016, 34, 337–344. [Google Scholar]
- Kampouris, E.M.; Papaspyrides, C.D.; Lekakou, C.N. A model recovery process for scrap polystyrene foam by means of solvent systems. Conserv. Recycl. 1987, 10, 315–319. [Google Scholar] [CrossRef]
- Kampouris, E.M.; Papaspyrides, C.D.; Lekakou, C.N. A model process for the solvent recycling of polystyrene. Polym. Eng. Sci. 1988, 28, 534–537. [Google Scholar] [CrossRef]
- Murugesh, A.K.; Uthayanan, A.; Lekakou, C. Electrophoresis and orientation of multiple wall carbon nanotubes in polymer solution. Appl. Phys. A 2010, 100, 135–144. [Google Scholar] [CrossRef]
- Huang, J.; Sumpter, B.G.; Meunier, V. Theoretical model for nanoporous carbon supercapacitors. Angew. Chem. Int. Ed. 2008, 47, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Mei, D.; Li, X.; Xu, W.; Wang, D.; Graff, G.L.; Bennett, W.D.; Nie, Z.; Saraf, L.V.; Aksay, I.A.; et al. Hierarchically porous graphene as a lithium-air battery electrode. Nano Lett. 2011, 11, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Mu, X.; Zheng, M.; Wang, C.; Lin, Z.; Zhang, X.; Shi, Y.; He, P.; Zhou, H. A multi-layered Fe2O3/graphene composite with mesopores as a catalyst for rechargeable aprotic lithium–oxygen batteries. Nanotechnology 2016, 27, 365402. [Google Scholar] [CrossRef]
- Shui, J.; Lin, Y.; Connell, J.W.; Xu, J.; Fan, X.; Dai, L. Nitrogen-doped holey graphene for high-performance rechargeable Li−O2 batteries. ACS Energy Lett. 2016, 1, 260–265. [Google Scholar] [CrossRef]
- Xu, G.; Ding, B.; Nie, P.; Shen, L.; Dou, H.; Zhang, X. Hierarchically porous carbon encapsulating sulfur as a superior cathode material for high performance lithium−sulfur batteries. ACS Appl. Mater. Interfaces 2014, 6, 194–199. [Google Scholar] [CrossRef]
- Robinson, J.; Xi, K.; Kumar, R.V.; Ferrari, A.C.; Au, H.; Titirici, M.M.; Puerto, A.P.; Kucernak, A.; Fitch, S.D.S.; Garcia-Araez, N. 2020 Roadmap on Lithium Sulfur Batteries. J. Phys. Energy 2021. accepted for publication. [Google Scholar]
- Lei, C.; Fields, R.; Wilson, P.; Lekakou, C.; Amini, N.; Tennison, S.; Perry, J.; Gosso, M.; Martorana, B. Development and evaluation of a composite supercapacitor-based 12 V transient start-stop (TSS) power system for vehicles: Modelling, design and fabrication scaling up. Proc. Inst. Mech. Eng. Part A J. Power Energy 2020. [Google Scholar] [CrossRef]
- Abouelamaiem, D.I.; He, G.; Neville, T.P.; Patel, D.; Ji, S.; Wang, R.; Parkin, I.P.; Jorge, A.B.; Titirici, M.-M.; Shearing, P.R.; et al. Correlating electrochemical impedance with hierarchical structure for porous carbon-based supercapacitors using a truncated transmission line model. Electrochim. Acta 2018, 284, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Huang, J.; Sumpter, B.G.; Meunier, V.; Qiao, R. Structure and dynamics of electrical double layers in organic electrolytes. Phys. Chem. Chem. Phys. 2010, 12, 5468–5479. [Google Scholar] [CrossRef] [PubMed]
- Babar, S.; Lekakou, C. Molecular modeling of electrolyte and polysulfide ions for lithium-sulfur batteries. Ionics 2021, 27, 635–642. [Google Scholar] [CrossRef]
- Lian, C.; Zhao, S.; Liu, H.; Wu, J. Time-dependent density functional theory for the charging kinetics of electric double layer containing room-temperature ionic liquids. J. Chem. Phys. 2016, 145, 204707. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, J.; Zhang, Z.; Wang, L.; Wang, W.; Liu, J.; Hong, Z.; Cho, K.; Wang, W. Origin of theoretical pseudocapacitance of two-dimensional supercapacitor electrodes Ti3C2T2 (T = bare, O, S). J. Mater. Chem. A 2019, 7, 16231–16238. [Google Scholar] [CrossRef]
- Drummond, R.; Howey, D.A.; Duncan, S.R. Low-order mathematical modelling of electric double layer supercapacitors using spectral methods. J. Power Sources 2015, 277, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Di Lecce, D.; Marangon, V.; Du, W.; Brett, D.J.L.; Shearing, P.R.; Hassoun, J. The role of synthesis pathway on the microstructural characteristics of sulfur-carbon composites: X-ray imaging and electrochemistry in lithium battery. J. Power Sources 2020, 472, 228424. [Google Scholar] [CrossRef]
- Bates, J. Modelling and Simulations of Energy Storage Devices. Ph.D. Thesis, University of Surrey, Guildford, UK, 2020. [Google Scholar]
- Markoulidis, F.; Bates, J.; Lekakou, C.; Slade, R.; Laudone, G.M. Supercapacitors with lithium-ion electrolyte: An experimental study and design of the activated carbon electrodes via modelling and simulations. Carbon 2020, 164, 422–434. [Google Scholar] [CrossRef]
- Bruggeman, D.A.G. Berechnung verschiedener physikalischer Konstanten von heterogenen Substanzen. I. Dielektrizitätskonstanten und Leitfähigkeiten der Mischkorper aus isotropen Substanzen. Ann. Phys. 1935, 416, 636–664. [Google Scholar] [CrossRef]
- Beck, R.E.; Schultz, J.S. Hindered diffusion in microporous membranes with known pore geometry. Science 1970, 170, 1302–1305. [Google Scholar] [CrossRef]
- Murch, G.E. The exact Nernst-Einstein equations and the interpretation of cross phenomenological coefficients in unary, binary, and ambipolar systems. Radiat. Eff. 1983, 73, 299–305. [Google Scholar] [CrossRef]
- Amico, S.; Lekakou, C. Flow through a two-scale porosity, oriented fibre porous medium. Transp. Porous Media 2004, 54, 35–53. [Google Scholar] [CrossRef]
- Lekakou, C.; Edwards, S.; Bell, G.; Amico, S.C. Computer modelling for the prediction of the in-plane permeability of non-crimp stitch bonded fabrics. Compos. A Appl. Sci. Manuf. 2006, 37, 820–825. [Google Scholar] [CrossRef]
- Elsayed, Y.; Lekakou, C.; Tomlins, P. Modeling, simulations and optimization of smooth muscle cell tissue engineering for the production of vascular grafts. Biotechnol. Bioeng. 2019, 116, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Lekakou, C.N.; Richardson, S.M. Simulation of reacting flow during filling in reaction injection molding (RIM). Polym. Eng. Sci. 1986, 26, 1264–1275. [Google Scholar] [CrossRef]
- Oldham, K.B. A Gouy–Chapman–Stern model of the double layer at a (metal)/(ionic liquid) interface. J. Electroanal. Chem. 2008, 613, 131–138. [Google Scholar]
- Kannan, D.; Terala, P.; Moss, P.; Weatherspoon, M. Analysis of the separator thickness and porosity on the performance of lithium-ion batteries. Int. J. Electrochem. 2018, 2018, 1925708. [Google Scholar] [CrossRef]
- Salifu, A.A.; Lekakou, C.; Labeed, F.H. Electrospun oriented gelatin-hydroxyapatite fiber scaffolds for bone tissue engineering. J. Biomed. Mater. Res. Part A 2017, 105, 1911–1926. [Google Scholar] [CrossRef]
- Salifu, A.A.; Nury, B.D.; Lekakou, C. Electrospinning of nanocomposite fibrillar tubular and flat scaffolds with controlled fiber orientation. Ann. Biomed. Eng. 2011, 39, 2510. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Zheng, W. A review for aqueous electrochemical supercapacitors. Front. Energy Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, G.Z. Ionic liquid-based electrolytes for supercapacitor and supercapattery. Front. Chem. 2019. [Google Scholar] [CrossRef]
- Heng, I.; Lai, C.W.; Juan, J.C. Organic electrolytes for supercapacitors. In Supercapacitor Technology; Materials Research Foundations: Millersville, PA, USA, 2019; Volume 61, pp. 1–10. [Google Scholar]
- Chmiola, J.; Largeot, C.; Taberna, P.-L.; Simon, P.; Gogotsi, Y. Desolvation of ions in subnanometer pores and its effect on capacitance and double-layer theory. Angew. Chem. Int. Ed. 2008, 47, 3392–3395. [Google Scholar] [CrossRef] [PubMed]
- Jagadale, A.; Zhou, X.; Xiong, R.; Dubal, D.P.; Xu, J.; Yang, S. Lithium ion capacitors (LICs): Development of the materials. Energy Storage Mater. 2019, 19, 314–329. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, T.; Yang, X.; Zhang, L.; Leng, K.; Huang, Y.; Chen, Y. A high-performance supercapacitor-battery hybrid energy storage device based on graphene-enhanced electrode materials with ultrahigh energy density. Energy Environ. Sci. 2013, 6, 1623–1632. [Google Scholar] [CrossRef]
- Choi, H.S.; Chong Park, R. Theoretical guidelines to designing high performance energy storage device based on hybridization of lithium-ion battery and supercapacitor. J. Power Sources 2014, 259, 1–14. [Google Scholar] [CrossRef]
- Vlad, A.; Singh, N.; Rolland, J.; Melinte, S.; Ajayan, P.M.; Gohy, J.-F. Hybrid supercapacitor-battery materials for fast electrochemical charge storage. Sci. Rep. 2014, 4, 4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Kawaharasaki, S.; Ito, K.; Yamada, S.; Hayamizub, K.; Sekic, S. Strategies for fast ion transport in electrochemical capacitor electrolytes from diffusion coefficients, ionic conductivity, viscosity, density and interaction energies based on HSAB theory. RSC Adv. 2017, 7, 14528–14535. [Google Scholar] [CrossRef] [Green Version]
- Perricone, E.; Chamas, M.; Cointeaux, L.; Leprêtre, J.-C.; Judeinstein, P.; Azais, P.; Béguin, F.; Alloin, F. Investigation of methoxypropionitrile as co-solvent for ethylene carbonate based electrolyte in supercapacitors. A safe and wide temperature range electrolyte. Electrochim. Acta 2013, 93, 1–7. [Google Scholar] [CrossRef]
- Logan, E.R.; Tonita, E.M.; Gering, K.L.; Li, J.; Ma, X.; Beaulieu, L.Y.; Dahn, J.R. A study of the physical properties of li-ion battery electrolytes containing esters. J. Electrochem. Soc. 2018, 165, A21–A30. [Google Scholar] [CrossRef]
- Afanas’ev, V.N.; Tyunina, E.Y.; Chekunova, M.D. The influence of temperature and concentration on viscous flow of solutions of Et4NBF4 in propylene carbonate. Russ. J. Phys. Chem. A 2009, 83, 2069–2073. [Google Scholar] [CrossRef]
- Markoulidis, F. Materials and Processing Techniques for the Fabrication of Supercapacitors for Automotive Applications. Ph.D. Thesis, University of Surrey, Guildford, UK, 2014. [Google Scholar]
- Hu, L.; Guo, D.; Feng, G.; Li, H.; Zhai, T. Asymmetric behavior of positive and negative electrodes in carbon/carbon supercapacitors and its underlying mechanism. J. Phys. Chem. C 2016, 120, 24675–24681. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Penner, R.M. Energy storage in nanomaterials—Capacitive, pseudocapacitive, or battery-like? ACS Nano 2018, 12, 2081–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forse, A.C.; Merlet, C.; Griffin, J.M.; Grey, C.P. New perspectives on the charging mechanisms of supercapacitors. J. Am. Chem. Soc. 2016, 138, 5731–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschamps, M.; Gilbert, E.; Azais, P.; Raymundo-Piñero, E.; Ramzi Ammar, M.; Simon, P.; Massiot, D.; Béguin, F. Exploring electrolyte organization in supercapacitor electrodes with solid-state NMR. Nat. Mater. 2013, 12, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Decaux, C.; Matei Ghimbeu, C.; Dahbi, M.; Anouti, M.; Lemordant, D.; Béguin, F.; Vix-Guterl, C.; Raymundo-Piñero, E. Influence of electrolyte ion–solvent interactions on the performances of supercapacitors porous carbon electrodes. J. Power Sources 2014, 263, 130–140. [Google Scholar] [CrossRef]









| Property | 1.5 M TEABF4/AN | 1.5 M TEABF4/PC | 1 M LiPF6/EC-EMC |
|---|---|---|---|
| kd | 0.876 | 0.6 | 1 |
| ηo (mPa s) | 0.37 | 1.8 | 1.4 |
| σ2 (S m−1) | 6 | 1.3 | 0.5 |
| Electrolyte Ion | Lmin,VdW (nm) 1 | nc 2 | Eion/solvent (kJ mol−1) 3 |
|---|---|---|---|
| TEA+ | 0.60 | 6(AN), 3(PC) | |
| BF4− | 0.45 | 3.3(AN), 3(PC) | |
| TEA+/AN | 1.1 | 15.5 | −222 |
| BF4−/AN | 0.85 | 6.75 | −196 |
| TEA+/PC | 1.28 | 13 | −10 |
| BF4−/PC | 1.04 | 6 | −35 |
| Li+ | 0.56 | 4 | |
| PF6− | 0.5 | 0 | |
| Li+/EC-EMC | 1.8 | 4 | −400 |
| PF6−/EC-EMC | 1.4 | 3 | −70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bates, J.; Markoulidis, F.; Lekakou, C.; Laudone, G.M. Design of Porous Carbons for Supercapacitor Applications for Different Organic Solvent-Electrolytes. C 2021, 7, 15. https://doi.org/10.3390/c7010015
Bates J, Markoulidis F, Lekakou C, Laudone GM. Design of Porous Carbons for Supercapacitor Applications for Different Organic Solvent-Electrolytes. C. 2021; 7(1):15. https://doi.org/10.3390/c7010015
Chicago/Turabian StyleBates, Joshua, Foivos Markoulidis, Constantina Lekakou, and Giuliano M. Laudone. 2021. "Design of Porous Carbons for Supercapacitor Applications for Different Organic Solvent-Electrolytes" C 7, no. 1: 15. https://doi.org/10.3390/c7010015
APA StyleBates, J., Markoulidis, F., Lekakou, C., & Laudone, G. M. (2021). Design of Porous Carbons for Supercapacitor Applications for Different Organic Solvent-Electrolytes. C, 7(1), 15. https://doi.org/10.3390/c7010015