NMR-Assisted Structure Elucidation of an Anticancer Steroid-β-Enaminone Derivative



Abstract
:
1. Introduction
2. Results and Discussion
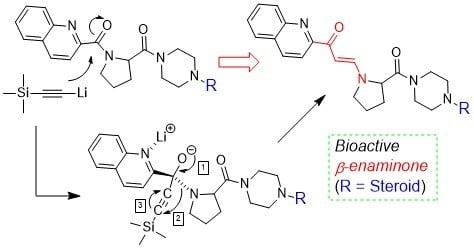
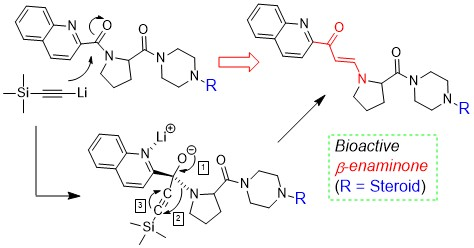
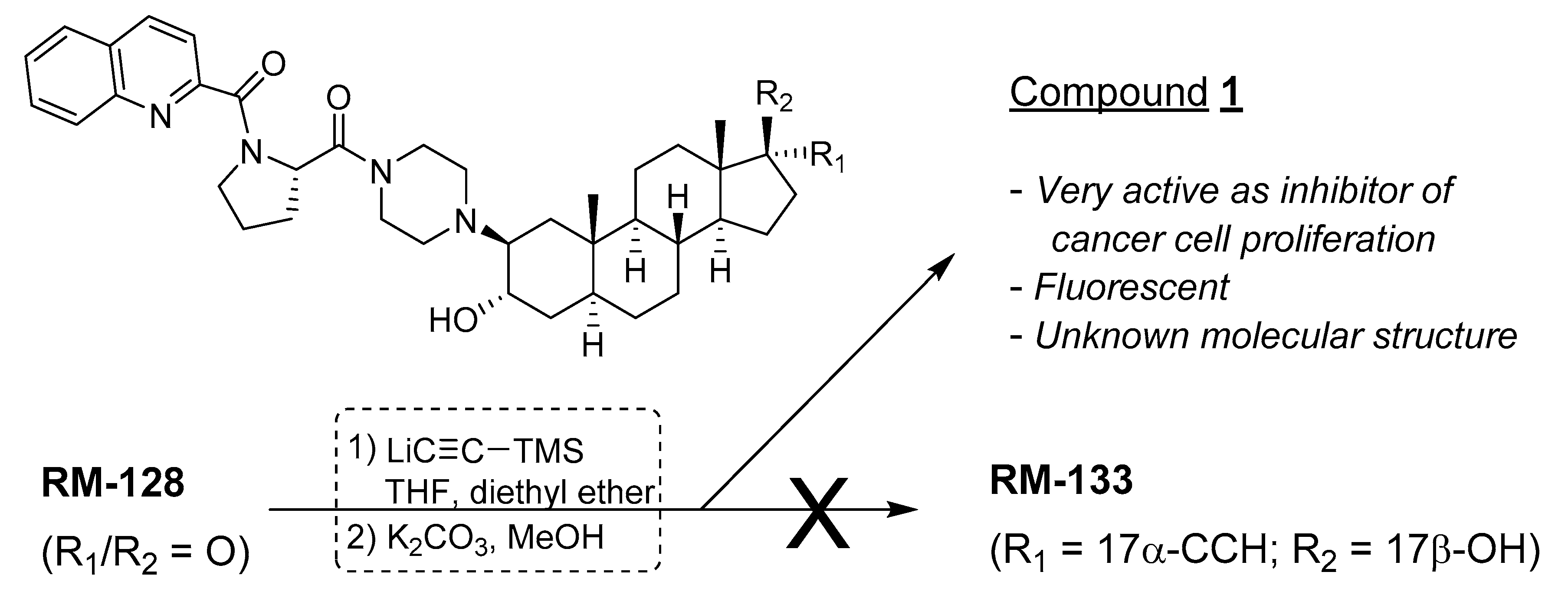
2.1. Characteristic of Unknown Compound and Hypothesis
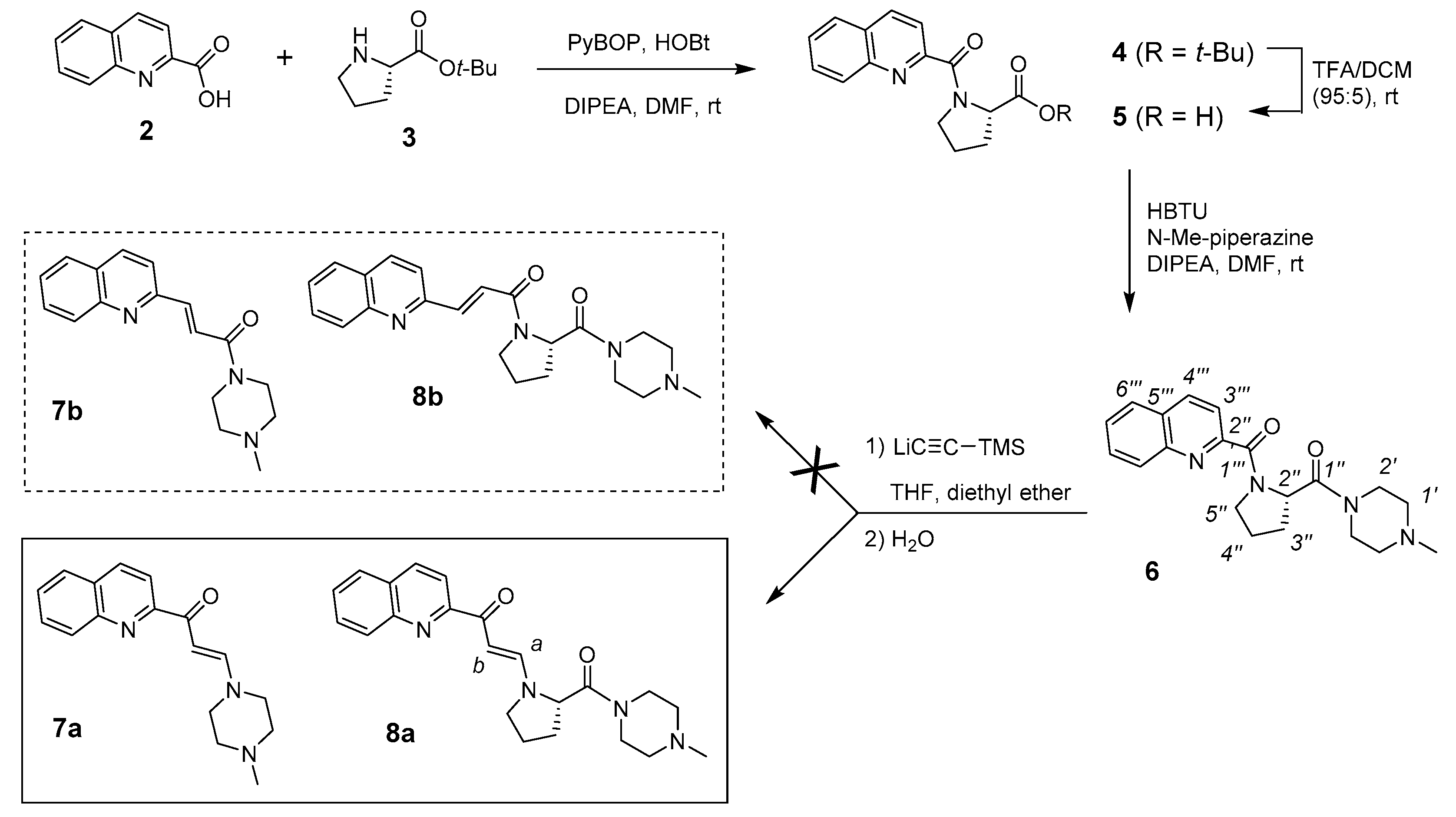
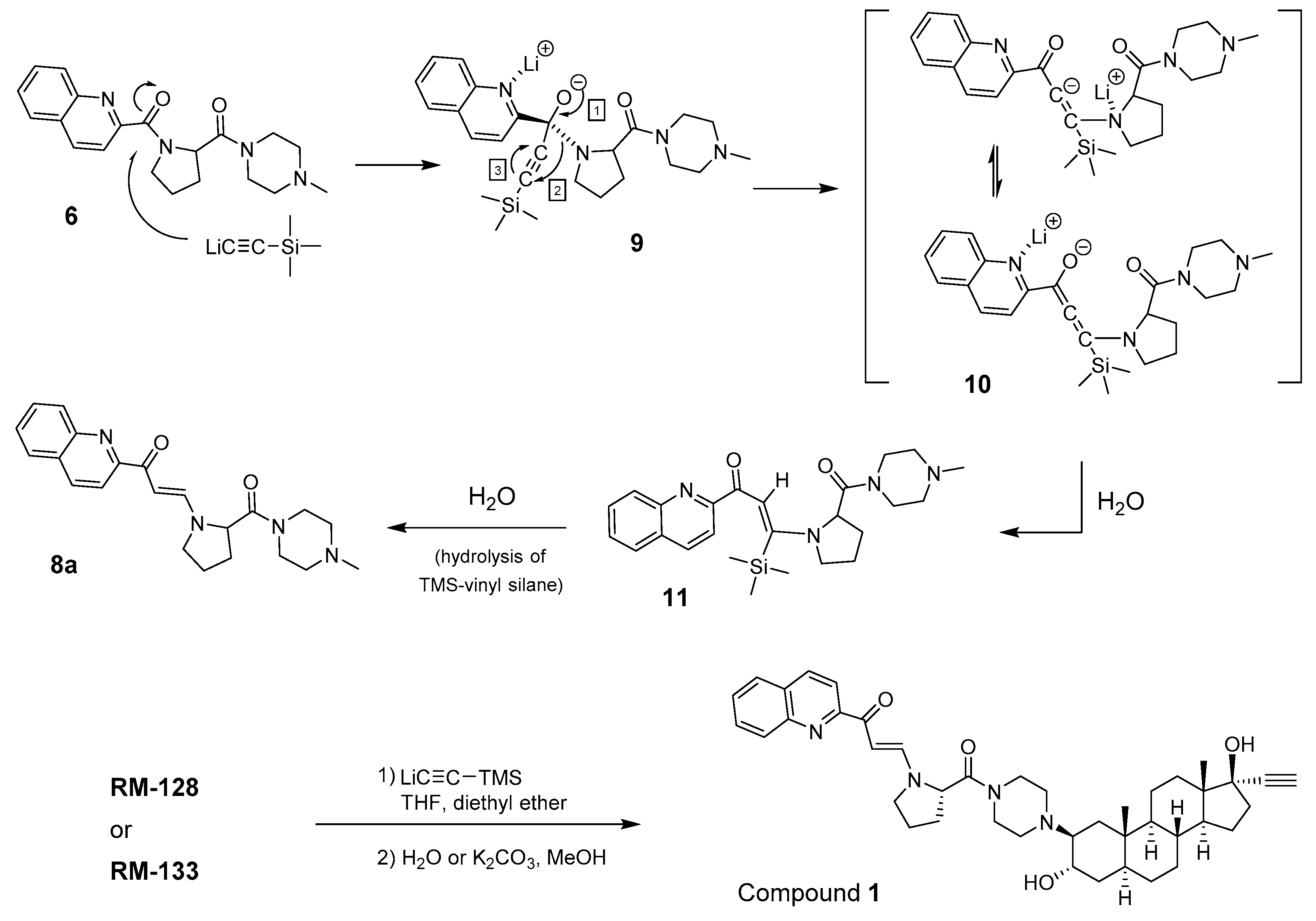
2.2. Synthesis of Side Chain 6 as a Model for the Formation of Unknown Compounds
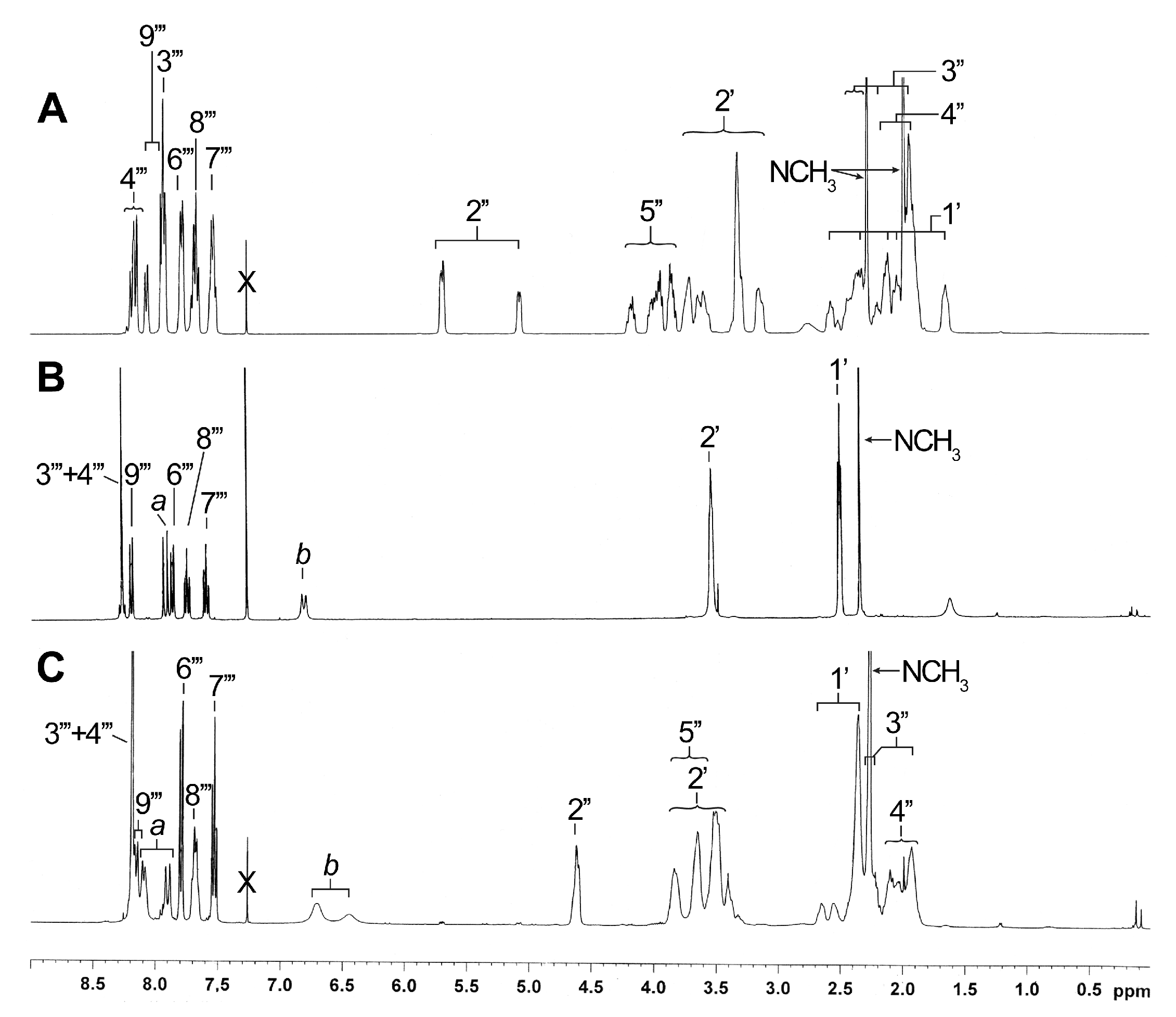
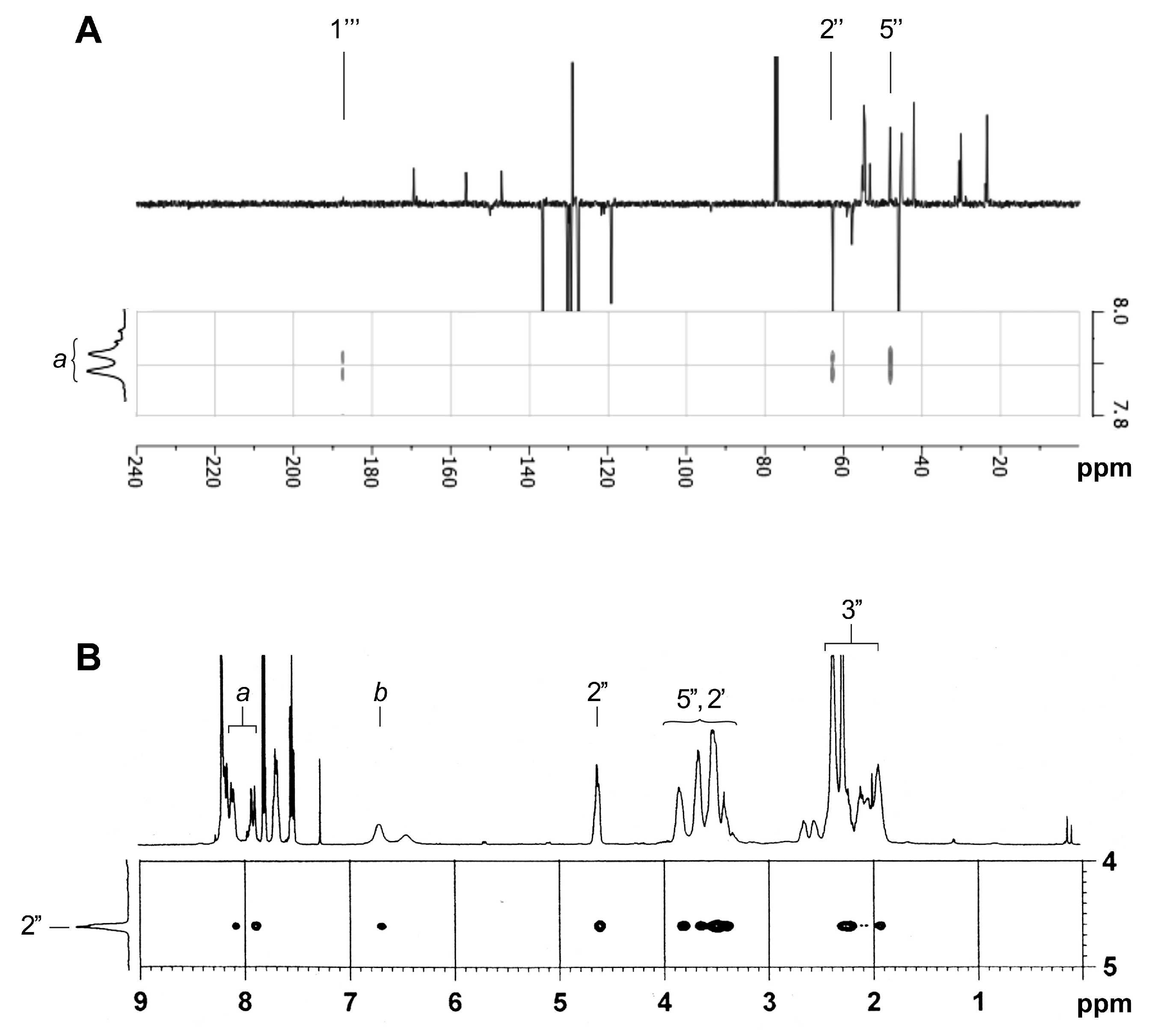
2.3. NMR Characterization of Unknown Side Chain (Enaminones 7 and 8)
2.4. NMR Characterization of Unknown Steroidal Compound 1
2.5. Mechanism of β-Enaminone Formation
3. Materials and Methods
3.1. General
3.2. Synthesis of Side Chain 6
3.3. Synthesis of β-Enaminones 7a and 8a
3.4. Synthesis of Enaminone 1
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, L.; Buoncervello, M.; Ascione, B.; Bellenghi, M.; Matarrese, P.; Care, A. The gender perspective in cancer research and therapy: Novel insights and on-going hypotheses. Ann. Ist. Super. Sanit. 2016, 52, 213–222. [Google Scholar]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.H.; Gershell, L.J. Selective anticancer drugs. Nat. Rev. Drug Discov. 2002, 1, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Maltais, R.; Roy, J.; Dutour, R.; Poirier, D. Design of a mestranol 2-N-piperazino-substituted derivative showing potent and selective in vitro and in vivo activities in MCF-7 breast cancer models. ChemMedChem 2017, 12, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Maltais, R.; Dutour, R.; Poirier, D. Explorative study on the anticancer activity, selectivity and metabolic stability of related analogs of aminosteroid RM-133. Steroids 2016, 115, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Maltais, R.; Hospital, A.; Delhomme, A.; Roy, J.; Poirier, D. Chemical synthesis, NMR analysis and evaluation on a cancer xenograft model (HL-60) of the aminosteroid derivative RM-133. Steroids 2014, 82, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ayan, D.; Maltais, R.; Hospital, A.; Poirier, D. Chemical synthesis, cytotoxicity, selectivity and bioavailability of 5alpha-androstane-3alpha,17beta-diol derivatives. Bioorg. Med. Chem. 2014, 22, 5847–5859. [Google Scholar] [CrossRef] [PubMed]
- Jegham, H.; Roy, J.; Maltais, R.; Desnoyers, S.; Poirier, D. A novel aminosteroid of the 5α-androstane-3α,17β-diol family induces cell cycle arrest and apoptosis in human promyelocytic leukemia HL-60 cells. Investig. New Drugs 2012, 30, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Jegham, H.; Maltais, R.; Roy, J.; Doillon, C.; Poirier, D. Biological evaluation of a new family of aminosteroids that display a selective toxicity for various malignant cell lines. Anti-Cancer Drugs 2012, 23, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kenmogne, L.C.; Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. The aminosteroid derivative RM-133 shows in vitro and in vivo antitumor activity in human ovarian and pancreatic cancers. PLoS ONE 2015, 10, e0144890. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.; Maltais, R.; Poirier, D. New diethylsilylacetylenic linker for parallel solid-phase synthesis of libraries of hydroxy acetylenic steroid derivatives with improved metabolic stability. ACS Comb. Sci. 2012, 14, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.M.; Ternes, T.A.; von Gunten, U. Removal of estrogenic activity and formation of oxidation products during ozonation of 17alpha-ethinylestradiol. Environ. Sci. Technol. 2004, 38, 5177–5186. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, K.L.; Cummings, R.I.; Hutchinson, T.H.; Scholze, M.; Brighty, G.; Sumpter, J.P.; Tyler, C.R. Relative potencies and combination effects of steroidal estrogens in fish. Environ. Sci. Technol. 2003, 37, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohkuma, T.; Tsuchihashi, G. Preparation of enaminones by two-carbon homologation of amides with lithium (triphenylsilyl)acetylide. J. Org. Chem. 1987, 52, 2929–2930. [Google Scholar] [CrossRef]
- Newman, H. Preparation of α,β-unsaturated aldehydes from acid chlorides. J. Org. Chem. 1973, 38, 2254–2255. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | 6 | 7a | 8a | 1 (Side Chain) | 1 (Steroid; C #) |
|---|---|---|---|---|---|
| CH2-4′′ | 22.3, 25.2 | -- | 23.4, 23.9 | 23.6, 23.9 | 12.9 (CH3-18) |
| CH2-3′′ | 28.8, 31.5 | -- | 30.1, 30.6 | 30.1, 30.7 | 17.2 (CH3-19) |
| CH2-2′ | 41.5, 42.0 44.7, 45.5 | 46.6 1 | 42.1, 45.1 45.5, 45.7 | 41.9, 42.7 45.8, 46.2 | 21.0 (CH2-11) |
| N-CH3 | 45.6, 45.9 | 46.0 | 46.0 | -- | 23.0 (CH2-15) |
| CH2-5′′ | 48.1, 49.8 | -- | 48.1, 53.3 | 48.1, 53.4 | 28.1 (CH2-6) |
| CH2-1′ | 54.2, 54.6, 54.9 | 54.6 1 | 54.5, 54.8 | 46.8, 48.0, 48.6 | 31.1 (CH2-7) |
| CH-2′′ | 57.4, 59.1 | -- | 57.9, 62.7 | 57.7, 62.7 | 32.7 (CH2-12) |
| CH-3′′′ | 120.9, 121.5 | 119.0 | 119.1 | 119.1 | 32.8 (CH2-1) |
| CH-7′′′ | 127.4 | 127.4 | 127.4 | 127.0 | 34.6 (CH2-4) |
| CH-6′′′ | 127.7 | 127.6 | 127.6 | 127.4 | 35.7 (C-10) |
| C-5′′′ | 128.1, 128.3 | 129.0 | 129.0 | 129.1 | 36.1 (CH-8) |
| CH-9′′′ | 129.2, 129.8 | 130.1 | 129.3, 130.3 | 129.9, 130.3 | 38.4 (CH-5) |
| CH-8′′′ | 129.6 | 129.4 | 129.4 | 129.4 | 38.9 (CH2-16) |
| CH-4′′′ | 136.6 | 136.6 | 136.5 | 136.5 | 47.0 (C-13) |
| C-10′′′ | 145.8, 146.3 | 147.0 | 147.1 | 147.1 | 50.3 (CH-14) |
| C-2′′′ | 153.4, 154.1 | 155.9 | 156.1 | 156.1 | 55.6 (CH-9) |
| C-1′′′ | 166.0, 166.6 | 187.6 | 187.42 | 187.5 | 63.7, 63.8 (CH-3) |
| C-1′′ | 169.0, 170.1 | -- | 168.6, 169.4 | 168.9, 169.2 | 64.8, 64.9 (CH-2) |
| CH-b | -- | 91.0 | 93.7 2 | 93.6 | 73.9 (C-20) |
| CH-a | -- | 153.0 | 150.0 2 | 149.7 | 79.8 (C-17) |
| -- | -- | -- | -- | -- | 87.6 (C-21) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poirier, D.; Maltais, R. NMR-Assisted Structure Elucidation of an Anticancer Steroid-β-Enaminone Derivative. Magnetochemistry 2017, 3, 37. https://doi.org/10.3390/magnetochemistry3040037
Poirier D, Maltais R. NMR-Assisted Structure Elucidation of an Anticancer Steroid-β-Enaminone Derivative. Magnetochemistry. 2017; 3(4):37. https://doi.org/10.3390/magnetochemistry3040037
Chicago/Turabian StylePoirier, Donald, and René Maltais. 2017. "NMR-Assisted Structure Elucidation of an Anticancer Steroid-β-Enaminone Derivative" Magnetochemistry 3, no. 4: 37. https://doi.org/10.3390/magnetochemistry3040037
APA StylePoirier, D., & Maltais, R. (2017). NMR-Assisted Structure Elucidation of an Anticancer Steroid-β-Enaminone Derivative. Magnetochemistry, 3(4), 37. https://doi.org/10.3390/magnetochemistry3040037