
Accidental Combustion Phenomena at Cryogenic Conditions



Abstract
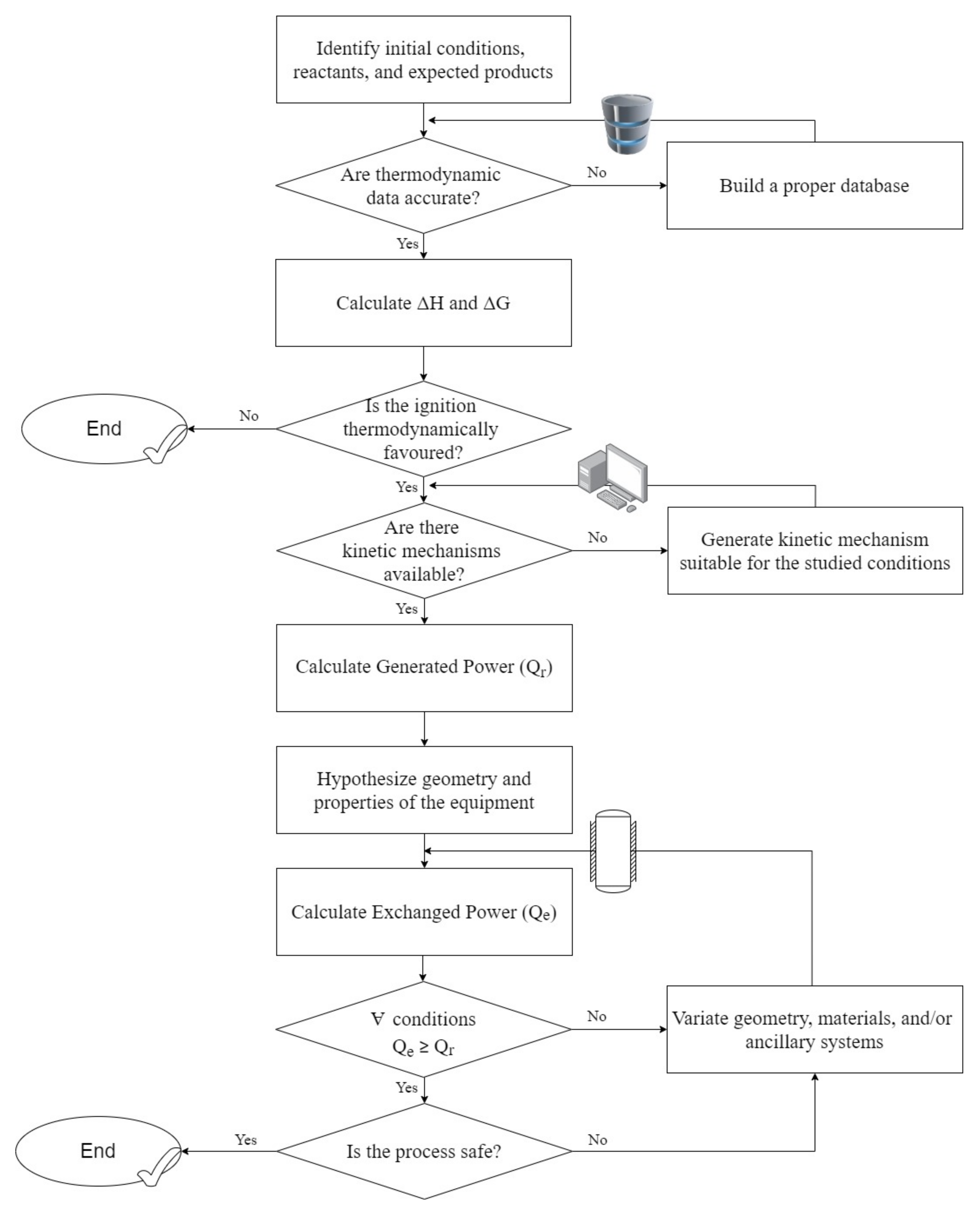
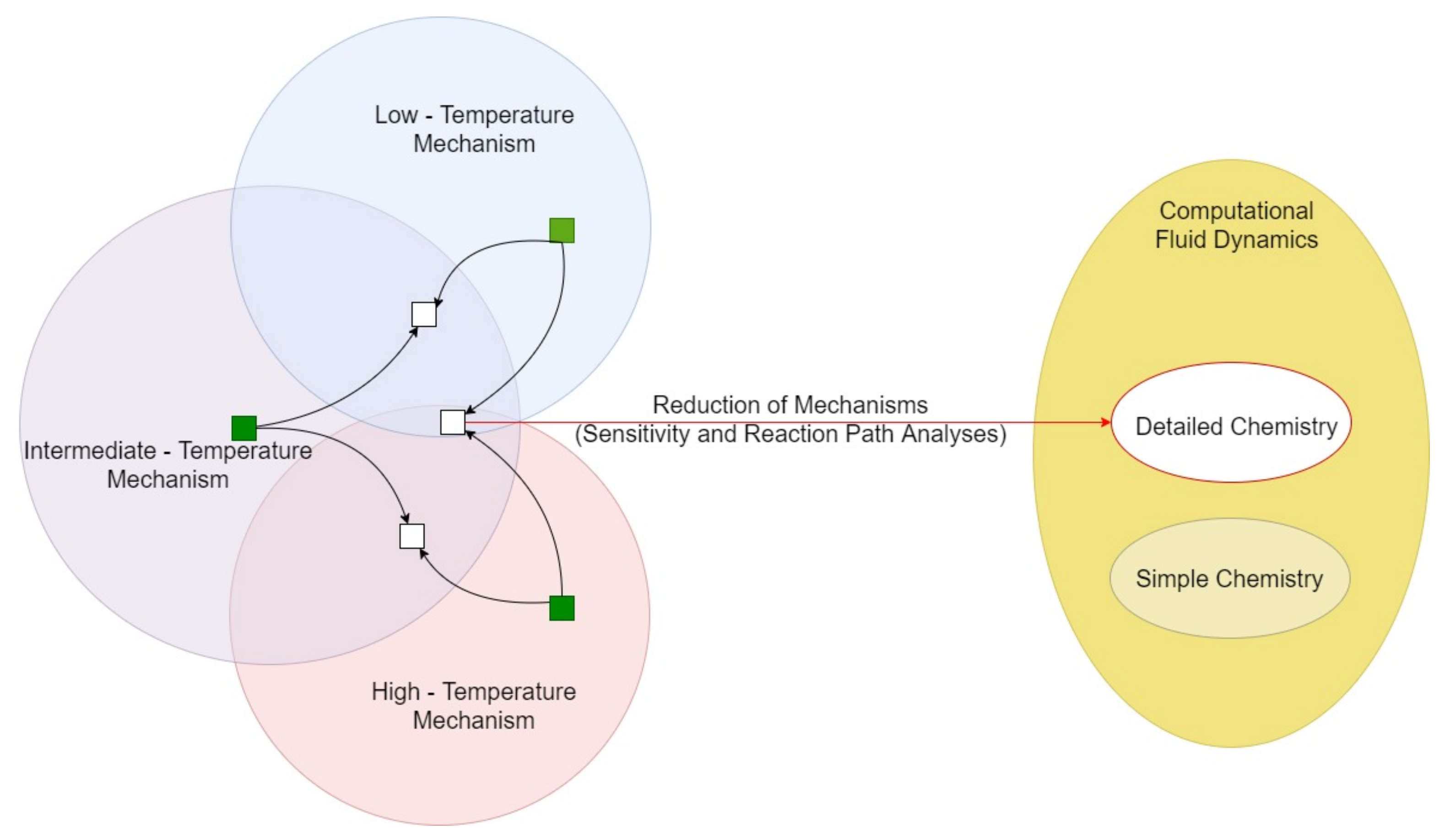
:1. Introduction
- -
- Laminar burning velocity
- -
- Flammability limits
- -
- Ignition (initiation reactions, ignition delay time)
- -
- Mass burning rate
- -
- Evaporation rate
2. Characterization of Gaseous Systems
2.1. Gaseous Properties
2.2. Laminar Burning Velocity
2.3. Flammability Limits
2.4. Ignition Phenomena
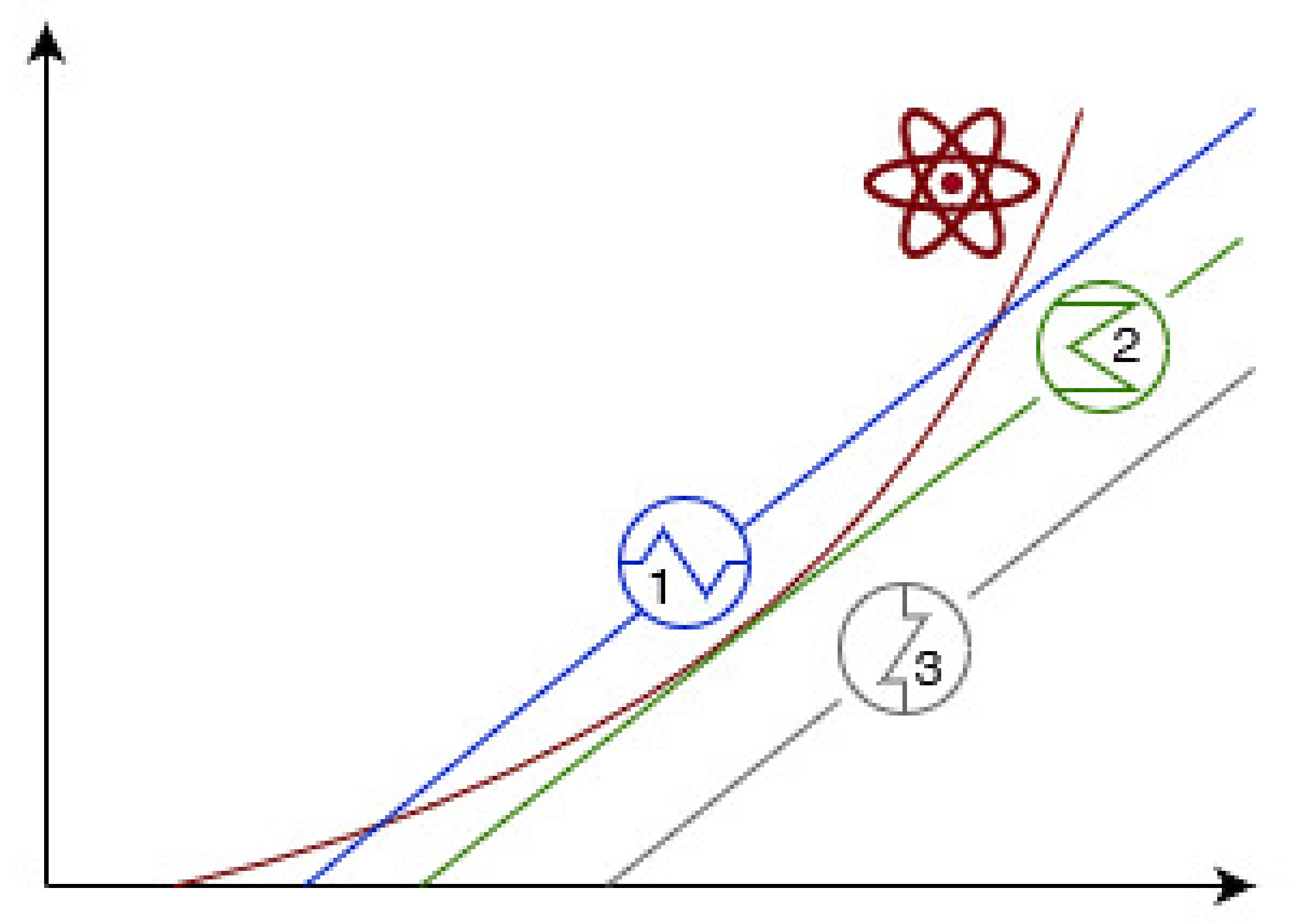
- Curves for produced heat and exchanged heat are secant. The conditions represented by the lower intersection are considered stable because the system can restore the initial conditions in case of moderate and localized variations. In other words, an increase in temperature leads to faster heat removal, with a consequent decrease of the bulk temperature, whereas a decrease in temperature makes heat removal slower than heat production, with a consequent increase of the bulk temperature. In contrast, the conditions representative for the intersection occurring at higher temperatures are considered unstable since any modification in operative temperature is accomplished by positive feedback.
- Curves for produced heat and exchanged heat are tangent. The conditions represented by the intersection behave like the ones corresponding to the intersection occurring at the higher temperature described in the previous case. Hence, they are intended as critical conditions.
- Curve for produced heat and exchanged heat are separated. The absence of intersections between the curves is representative for super-critical conditions, thus to unstable points regardless of the temperature variation.
3. Characterization of Liquid-Vapor Systems
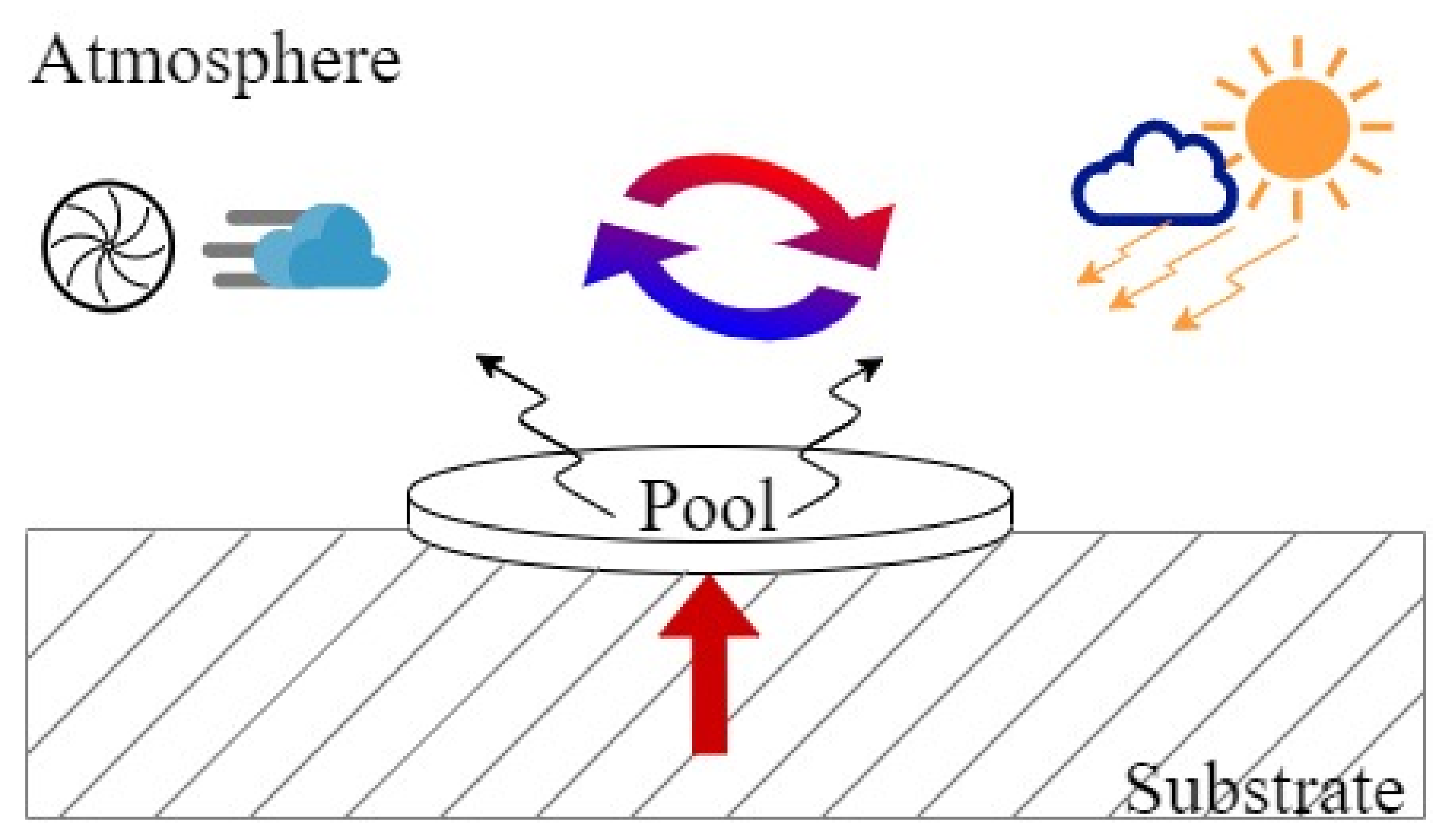
3.1. Evaporating Pool
- condensation of evaporated molecules in the area surrounding the liquid.
- evaporation of liquid molecules in the atmosphere.
3.2. Non-Reactive Liquid-Vapor Systems
3.3. Reactive Liquid-Vapor Systems
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Berstad, D.O.; Stang, J.H.; Nekså, P. Comparison criteria for large-scale hydrogen liquefaction processes. Int. J. Hydrogen Energy 2009, 34, 1560–1568. [Google Scholar] [CrossRef]
- McQuarrie, D.A.; Simon, J.D. Physical Chemistry—A Molecular Approach, 2nd ed.; University Science Books: Sausalito, CA, USA, 1997. [Google Scholar] [CrossRef]
- Razus, D.; Movileanu, C.; Brinzea, V.; Oancea, D. Explosion pressures of hydrocarbon-air mixtures in closed vessels. J. Hazard. Mater. 2006, 135, 58–65. [Google Scholar] [CrossRef] [PubMed]
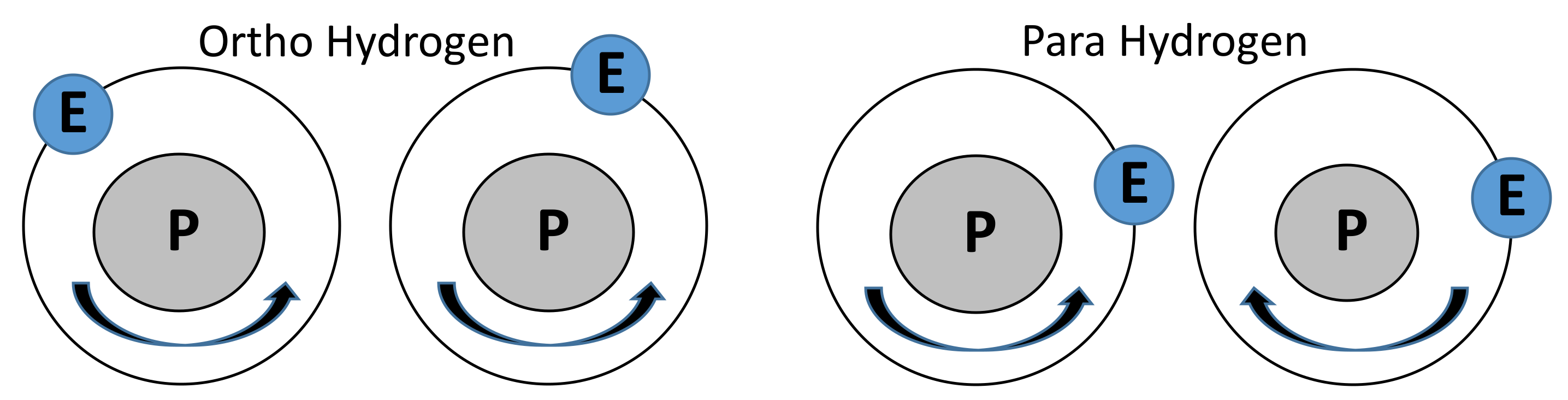
- Colonna, G.; D’Angola, A.; Capitelli, M. Statistical thermodynamic description of H2 molecules in normal ortho/para mixture. Int. J. Hydrogen Energy 2012, 37, 9656–9668. [Google Scholar] [CrossRef]
- Giauque, F.W. The Entropy of Hydrogen and the Third Law of Thermodynamics the Free Energy and Dissociation of Hydrogen. Contrib. Chem. Lab. Univ. Calif. 1930, 52, 4816–4831. [Google Scholar] [CrossRef]
- Cook, D.B. Ab initio valence calculations in chemistry. J. Mol. Struct. 1976. [Google Scholar] [CrossRef]
- Pounds, A.J. Introduction to Quantum Mechanics: A Time-Dependent Perspective (David J. Tannor), 1st ed.; University Science Books: Sausalito, CA, USA, 2007. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Musyoka, N.M.; Langmi, H.W.; Mathe, M.; Liao, S. Current research trends and perspectives on materials-based hydrogen storage solutions: A critical review. Int. J. Hydrogen Energy 2016, 42, 289–311. [Google Scholar] [CrossRef]
- Gillis, R.C.; Bailey, T.; Gallmeier, F.X.; Hartl, M.A. Raman Spectroscopy as an ortho-para diagnostic of liquid hydrogen moderators. J. Phys. 2018, 1021, 1–4. [Google Scholar] [CrossRef]
- Woolley, B.H.W.; Scott, R.B.; Brickwedde, F.G. Compilation of Thermal Properties of Hydrogen in Its Various Isotopic and Ortho-Para Modifications. J. Res. Natl. Bur. Stand. 1948, 41, 379–475. [Google Scholar] [CrossRef]
- Jensen, J.E.; Tuttle, W.A.; Stewart, R.B.; Brechna, H.; Prodell, A.G. Cryogenic Data Handbook; Brookhaven National Laboratory: New York, NY, USA, 1980.
- Le Roy, R.J.; Chapman, S.G.; McCourt, F.R.W. Accurate thermodynamic properties of the six isotopomers of diatomic hydrogen. J. Phys. Chem. 1990, 94, 923–929. [Google Scholar] [CrossRef]
- Li, L.; Xie, M.; Wei, W.; Jia, M.; Liu, H. Numerical investigation on cryogenic liquid jet under transcritical and supercritical conditions. Cryogenics 2018, 89, 16–28. [Google Scholar] [CrossRef]
- Ancheyta, J.; Sánchez, S.; Rodríguez, M.A. Kinetic modeling of hydrocracking of heavy oil fractions: A review. Catal. Today 2005, 109, 76–92. [Google Scholar] [CrossRef]
- Egolfopoulos, F.N.; Hansen, N.; Ju, Y.; Kohse-Hoinghaus, K.; Law, C.K.; Qi, F. Advances and challenges in laminar flame experiments and implications for combustion chemistry. Prog. Energy Combust. Sci. 2014, 43, 36–67. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. Prog. Energy Combust. Sci. 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Curran, H.J. Developing detailed chemical kinetic mechanisms for fuel combustion. Proc. Combust. Inst. 2019, 37, 57–81. [Google Scholar] [CrossRef]
- Vandewiele, N.M.; Van Geem, K.M.; Reyniers, M.F.; Marin, G.B. Genesys: Kinetic model construction using chemo-informatics. Chem. Eng. J. 2012. [Google Scholar] [CrossRef]
- Gao, C.W.; Allen, J.W.; Green, W.H.; West, R.H. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 2016, 203, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Glassman, I.; Yetter, R.A. Combustion; Springer: Berlin, Germany, 2008. [Google Scholar] [CrossRef]
- Burcat, A.; Branko, R. Third Millennium Ideal Gas and Condensed Phase Thermochemical Database for Combustion with Updates from Active Thermochemical Tables; Technical Report; Argonne National Laboratory (ANL): Lemont, IL, USA, 2005. [CrossRef] [Green Version]
- Cohen, N. Revised group additivity values for enthalpies of formation (at 298 K) of carbon-hydrogen and carbon-hydrogen-oxygen compounds. J. Phys. Chem. Ref. Data 1996, 25, 1411–1483. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sheen, D.A. Combustion kinetic model uncertainty quantification, propagation and minimization. Prog. Energy Combust. Sci. 2015, 47, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Zádor, J.; Klippenstein, S.J.; Miller, J.A. Pressure-dependent OH yields in alkene + HO2 reactions: A theoretical study. J. Phys. Chem. A 2011, 115, 10218–10225. [Google Scholar] [CrossRef] [PubMed]
- Glarborg, P.; Miller, J.A.; Ruscic, B.; Klippenstein, S.J. Modeling nitrogen chemistry in combustion. Prog. Energy Combust. Sci. 2018, 67, 31–68. [Google Scholar] [CrossRef] [Green Version]
- Benson, S.W. Thermochemical Kinetics: Methods for the Estimation of Thermochemical Data and Rate Parameters. In Thermochemical Kinetics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1976; ISBN 0471067814. [Google Scholar] [CrossRef]
- Dong, X.; Ninnemann, E.; Ranasinghe, D.S.; Laich, A.; Greene, R.; Vasu, S.S.; Green, W.H. Revealing the critical role of radical-involved pathways in high temperature cyclopentanone pyrolysis. Combust. Flame 2020, 216, 280–292. [Google Scholar] [CrossRef]
- Bachrach, S.M. Organic Chemistry Computational, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; ISBN 9781118291924. [Google Scholar]
- Somers, K.P.; Simmie, J.M. Benchmarking Compound Methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the Active Thermochemical Tables: Formation Enthalpies of Radicals. J. Phys. Chem. A 2015, 119, 8922–8933. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N.; Klippenstein, S.J.; Taatjes, C.A.; Miller, J.A.; Wang, J.; Cool, T.A.; Yang, B.; Yang, R.; Wei, L.; Huang, C.; et al. Identification and chemistry of C 4H 3 and C 4H 5 isomers in fuel-rich flames. J. Phys. Chem. A 2006, 110, 3670–3678. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; Von Lilienfeld, O.A. Big data meets quantum chemistry approximations: The Δ-machine learning approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Ochterski, J.W.; Petersson, G.A.; Montgomery, J.A. A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar] [CrossRef]
- Pickard, F.C.; Pokon, E.K.; Liptak, M.D.; Shields, G.C. Comparison of CBS-QB3, CBS-APNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. J. Chem. Phys. 2005, 122, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zádor, J.; Taatjes, C.A.; Fernandes, R.X. Kinetics of elementary reactions in low-temperature autoignition chemistry. Prog. Energy Combust. Sci. 2011, 37, 371–421. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1070–1101. [Google Scholar] [CrossRef]
- Atkin, P.; Paula, J. Physical Chemistry; Royal Society of Chemistry: London, UK, 2006; ISBN 978-0716787594. [Google Scholar] [CrossRef]
- Battin-Leclerc, F.; Simmie, J.M.; Blurock, E. Cleaner Combustion—Developing Detailed Chemical Kinetic Models; Springer: Berlin/Heidelberg, Germany, 2003; ISBN 9781447153061. [Google Scholar] [CrossRef] [Green Version]
- Salzano, E.; Carboni, M.; Pio, G. The effects of low-temperature phenomena on rapid phase transition of liquid hydrogen. Int. J. Hydrogen Energy 2020, 45, 32676–32685. [Google Scholar] [CrossRef]
- Carboni, M.; Pio, G.; Vianello, C.; Maschio, G.; Salzano, E. Large eddy simulation for the rapid phase transition of LNG. Saf. Sci. 2021, 133, 105001. [Google Scholar] [CrossRef]
- Saggese, C.; Frassoldati, A.; Cuoci, A.; Faravelli, T.; Ranzi, E. A wide range kinetic modeling study of pyrolysis and oxidation of benzene. Combust. Flame 2013, 160, 1168–1190. [Google Scholar] [CrossRef]
- Goldsborough, S.S.; Hochgreb, S.; Vanhove, G.; Wooldridge, M.S.; Curran, H.J.; Sung, C.J. Advances in rapid compression machine studies of low- and intermediate-temperature autoignition phenomena. Prog. Energy Combust. Sci. 2017, 63, 1–78. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.K.; Singh, A.P.; Maurya, R.K. Evolution, challenges and path forward for low temperature combustion engines. Prog. Energy Combust. Sci. 2017, 61, 1–56. [Google Scholar] [CrossRef]
- Lele, A.D.; Vallabhuni, S.K.; Moshammer, K.; Fernandes, R.X.; Krishnasamy, A.; Narayanaswamy, K. Experimental and chemical kinetic modeling investigation of methyl butanoate as a component of biodiesel surrogate. Combust. Flame 2018, 197, 49–64. [Google Scholar] [CrossRef]
- Bhaskaran, K.A.; Roth, P. The shock tube as wave reactor for kinetic studies and material systems. Prog. Energy Combust. Sci. 2002, 28, 151–192. [Google Scholar] [CrossRef]
- Hertzberg, M. The Theory of Flammability Limits, Flow Gradient Effect and Flame Stretch; US Department of the Interior, Bureau of Mines: Washington, DC, USA, 1984.
- Shy, S.S.; Lin, W.J.; Wei, J.C. An experimental correlation of turbulent burning velocities for premixed turbulent methane-air combustion. Proc. R. Soc. A Math. Phys. Eng. Sci. 2000, 456, 1997–2019. [Google Scholar] [CrossRef]
- Kelley, A.P.; Law, C.K. Nonlinear effects in the extraction of laminar flame speeds from expanding spherical flames. Combust. Flame 2009, 156, 1844–1851. [Google Scholar] [CrossRef]
- Dowdy, D.R.; Smith, D.B.; Taylor, S.C.; Williams, A. The use of expanding spherical flames to determine burning velocities and stretch effects in hydrogen/air mixtures. Symp. Combust. 1991, 23, 325–332. [Google Scholar] [CrossRef]
- Mcenally, C.S.; Pfefferle, L.D.; Atakan, B.; Kohse-Höinghaus, K.; Kohse-ho, K. Studies of aromatic hydrocarbon formation mechanisms in flames: Progress towards closing the fuel gap. Prog. Energy Combust. Sci. 2006, 32, 247–294. [Google Scholar] [CrossRef]
- De Goey, L.P.H.; van Maaren, A.; Ouax, R.M. Stabilization of Adiabatic Premixed Laminar Flames on a Flat Flame Burner. Combust. Sci. Technol. 1993, 92, 201–207. [Google Scholar] [CrossRef]
- Tran, L.-S.; Wullenkord, J.; Li, Y.; Herbinet, O.; Zeng, M.; Qi, F.; Kohse-Höinghaus, K.; Battin-Leclerc, F. Probing the low-temperature chemistry of di-n-butyl ether: Detection of previously unobserved intermediates. Combust. Flame 2019, 210, 9–24. [Google Scholar] [CrossRef]
- Dayma, G.; Thion, S.; Lailliau, M.; Serinyel, Z.; Dagaut, P.; Sirjean, B.; Fournet, R. Kinetics of propyl acetate oxidation: Experiments in a jet-stirred reactor, ab initio calculations, and rate constant determination. Proc. Combust. Inst. 2019, 37, 429–436. [Google Scholar] [CrossRef]
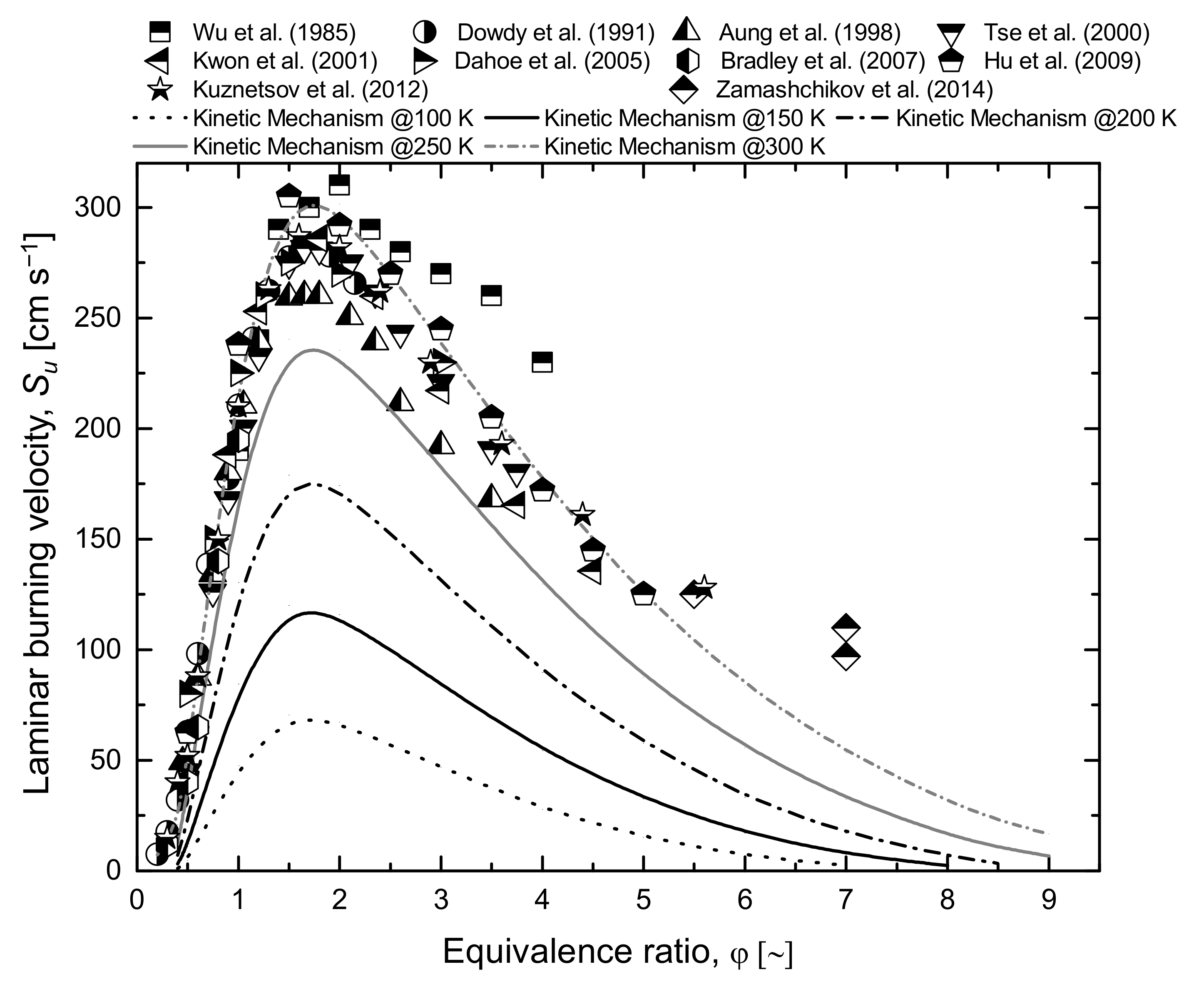
- Wu, C.K.; Law, C.K. On the determination of laminar flame speeds from stretched flames. Symp. Combust. 1985, 20, 1941–1949. [Google Scholar] [CrossRef] [Green Version]
- Aung, K.T.; Hassan, M.I.; Faeth, G.M. Effects of pressure and nitrogen dilution on flame stretch interactions of laminar premixed H2O2 N2 flames. Combust. Flame 1998, 112, 1–15. [Google Scholar] [CrossRef]
- Tse, S.D.; Zhu, D.L.; Law, C.K. Morphology and burning rates of expanding spherical flames in H2/O2/inert mixtures up to 60 atmospheres. Proc. Combust. Inst. 2000, 28, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Kwon, O.C.; Faeth, G.M. Flame/stretch interactions of premixed hydrogen-fueled flames: Measurements and predictions. Combust. Flame 2001, 124, 590–610. [Google Scholar] [CrossRef]
- Dahoe, A.E. Laminar burning velocities of hydrogen-air mixtures from closed vessel gas explosions. J. Loss Prev. Process Ind. 2005, 18, 152–166. [Google Scholar] [CrossRef]
- Bradley, D.; Lawes, M.; Liu, K.; Verhelst, S.; Woolley, R. Laminar burning velocities of lean hydrogen-air mixtures at pressures up to 1.0 MPa. Combust. Flame 2007, 149, 162–172. [Google Scholar] [CrossRef]
- Hu, E.; Huang, Z.; He, J.; Miao, H. Experimental and numerical study on laminar burning velocities and flame instabilities of hydrogen-air mixtures at elevated pressures and temperatures. Int. J. Hydrogen Energy 2009, 34, 8741–8755. [Google Scholar] [CrossRef]
- Kuznetsov, M.; Czerniak, M.; Grune, J.; Jordan, T. Effect of Temperature on Laminar Flame Velocity for Hydrogen-Air Mixtures At Reduced Pressures. In Proceedings of the International Conference of Hydrogen Safety, Brussels, Belgium, 9–11 September 2013; pp. 1–12. [Google Scholar]
- Zamashchikov, V.V.; Alekseev, V.A.; Konnov, A.A. Laminar burning velocities of rich near-limiting flames of hydrogen. Int. J. Hydrogen Energy 2014, 39, 1874–1881. [Google Scholar] [CrossRef]
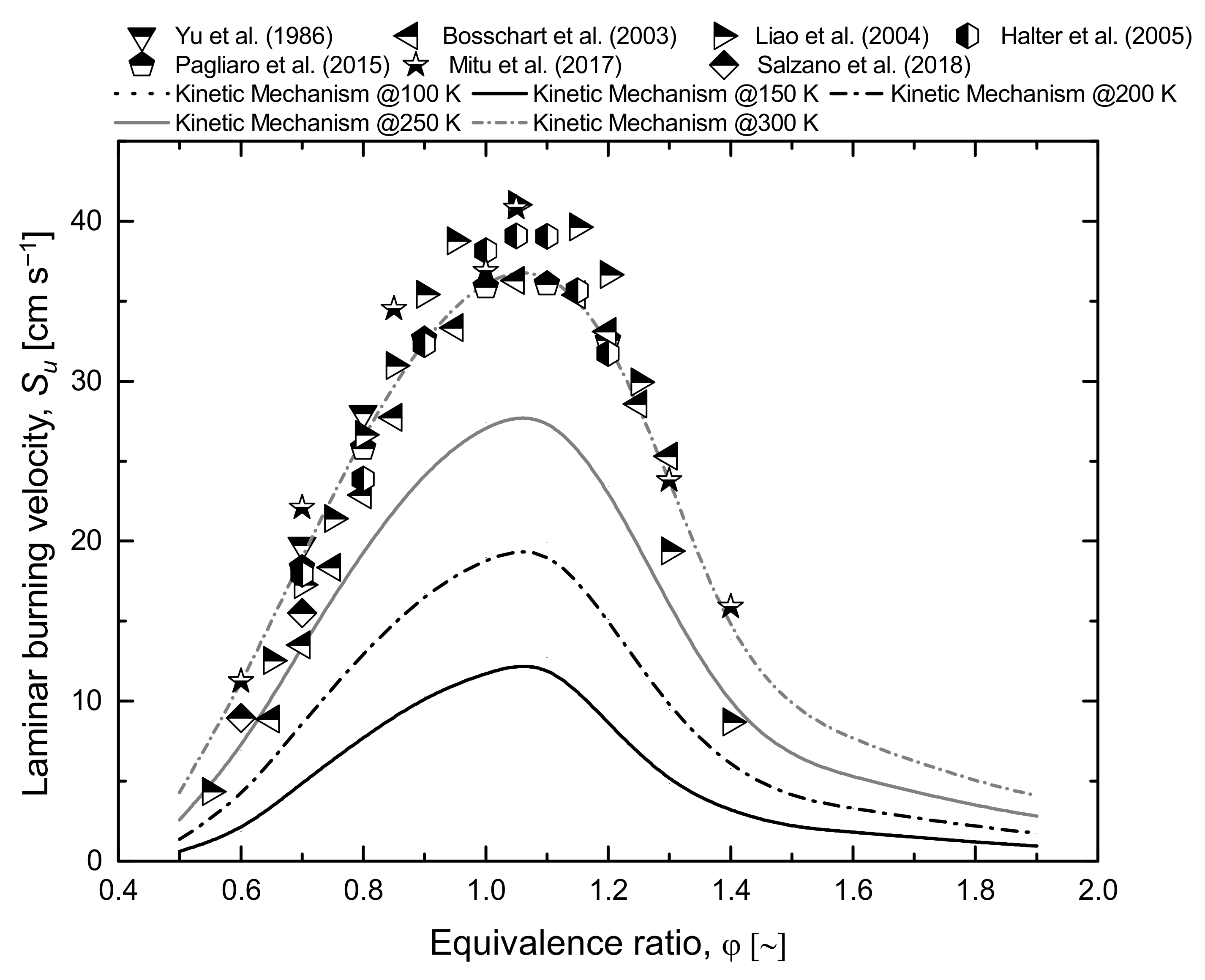
- Yu, G.; Law, C.K.; Wu, C.K. Laminar flame speeds of hydrocarbon + air mixtures with hydrogen addition. Combust. Flame 1986, 63, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Bosschaart, K.J.; de Goey, L.P.H. Detailed analysis of the heat flux method for measuring burning velocities. Combust. Flame 2003, 132, 170–180. [Google Scholar] [CrossRef]
- Liao, S.Y.; Jiang, D.M.; Cheng, Q. Determination of laminar burning velocities for natural gas. Fuel 2004, 83, 1247–1250. [Google Scholar] [CrossRef]
- Halter, F.; Chauveau, C.; Djebaïli-Chaumeix, N.; Gökalp, I. Characterization of the effects of pressure and hydrogen concentration on laminar burning velocities of methane-hydrogen-air mixtures. Proc. Combust. Inst. 2005, 30, 201–208. [Google Scholar] [CrossRef]
- Pagliaro, J.L.; Linteris, G.T.; Sunderland, P.B.; Baker, P.T. Combustion inhibition and enhancement of premixed methane-air flames by halon replacements. Combust. Flame 2015, 162, 41–49. [Google Scholar] [CrossRef]
- Mitu, M.; Giurcan, V.; Razus, D.; Oancea, D. Inert gas influence on the laminar burning velocity of methane-air mixtures. J. Hazard. Mater. 2017, 321, 440–448. [Google Scholar] [CrossRef]
- Salzano, E.; Pio, G.; Ricca, A.; Palma, V. The effect of a hydrogen addition to the premixed flame structure of light alkanes. Fuel 2018, 234, 1064–1070. [Google Scholar] [CrossRef]
- Pio, G.; Barba, D.; Palma, V.; Salzano, E. A Numerical Study on the Effect of Temperature and Composition on the Flammability of Methane–Hydrogen Sulfide Mixtures. Combust. Sci. Technol. 2019, 191, 1541–1557. [Google Scholar] [CrossRef]
- Coward, H.F.; Jones, G.W. Bulletin 508: Limits of Flammability of Gases and Vapors; Springfield: Geneseo, IL, USA, 1952. [Google Scholar] [CrossRef] [Green Version]
- Le, H.; Nayak, S.; Mannan, M.S. Upper flammability limits of hydrogen and light hydrocarbons in air at subatmospheric pressures. Ind. Eng. Chem. Res. 2012, 51, 9396–9402. [Google Scholar] [CrossRef]
- Van Den Schoor, F.; Verplaetsen, F. The upper explosion limit of lower alkanes and alkenes in air at elevated pressures and temperatures. J. Hazard. Mater. 2006. [Google Scholar] [CrossRef]
- Crowl, D.A.; Jo, Y. Do A method for determining the flammable limits of gases in a spherical vessel. Process Saf. Prog. 2009, 28, 227–236. [Google Scholar] [CrossRef]
- ASTM. Standard Test Method for Concentration Limits of Flammability of Chemicals (Vapors and Gases) 1; ASTM: West Conshohocken, PA, USA, 2009. [Google Scholar] [CrossRef]
- CEN EN 1839 Determination of the Explosion Limits and the Limiting Oxygen Concentration(LOC) for Flammable Gases and Vapours—IHS Engineering Workbench. 2017. Available online: https://ewb.ihs.com/#/document/ETYYSFAAAAAAAAAA?qid=636471168940254100&sr=re-1-10&kbid=4%7C20027&docid=942576196#h319b0c22 (accessed on 10 May 2021).
- Molnarne, M.; Schroeder, V. Flammability of gases in focus of European and US standards. J. Loss Prev. Process Ind. 2017, 48, 297–304. [Google Scholar] [CrossRef]
- Zabetakis, M.G. Flammability Characteristics of Combustible Gases and Vapors; U. S. Government Printing Office: Washington, DC, USA, 1965.
- Adler, J.; Enig, J.W. The critical conditions in thermal explosion theory with reactant consumption. Combust. Flame 1964, 8, 97–103. [Google Scholar] [CrossRef]
- Li, R.; Liu, Z.; Han, Y.; Tan, M.; Xu, Y.; Tian, J.; Chai, J.; Liu, J. Extended adiabatic flame temperature method for lower flammability limits prediction of fuel-air-diluent mixture by nonstoichiometric equation and nitrogen equivalent coefficients. Energy Fuels 2017, 31, 351–361. [Google Scholar] [CrossRef]
- Razus, D.; Molnarne, M.; Fuß, O. Limiting oxygen concentration evaluation in flammable gaseous mixtures by means of calculated adiabatic flame temperatures. Chem. Eng. Process. Process Intensif. 2004, 43, 775–784. [Google Scholar] [CrossRef]
- Karim, G.A.; Wierzba, I.; Boon, S. The lean flammability limits in air of methane, hydrogen and carbon monoxide at low temperatures. Cryogenics 1984, 24, 305–308. [Google Scholar] [CrossRef]
- Wierzba, I.; Wang, Q. The flammability limits of H2–CO–CH4 mixtures in air at elevated temperatures. Int. J. Hydrogen Energy 2006, 31, 485–489. [Google Scholar] [CrossRef]
- Siu, N.; Herring, J.S.; Byers, J. Qualitative Risk Assessment for an LNG Refueling Station ’ and Review of Relevant Safety Issues; Idaho National Engineering Lab.: Idaho Falls, ID, USA, 1999.
- Li, Z.; Gong, M.; Sun, E.; Wu, J.; Zhou, Y. Effect of low temperature on the flammability limits of methane/nitrogen mixtures. Energy 2011, 36, 5521–5524. [Google Scholar] [CrossRef]
- Cui, G.; Li, Z.; Yang, C. Experimental study of flammability limits of methane/air mixtures at low temperatures and elevated pressures. Fuel 2016, 181, 1074–1080. [Google Scholar] [CrossRef]
- Pio, G.; Ricca, A.; Palma, V.; Salzano, E. Flammability limits of methane/alkene mixtures in air. Chem. Eng. Trans. 2019, 77, 169–174. [Google Scholar] [CrossRef]
- Carriere, T.; Westmoreland, P.R.; Kazakov, A.; Stein, Y.S.; Dryer, F.L. Modeling ethylene combustion from low to high pressure. Proc. Combust. Inst. 2002, 29, 1257–1266. [Google Scholar] [CrossRef]
- Muñoz, M.; Planas, E.; Ferrero, F.; Casal, J. Predicting the emissive power of hydrocarbon pool fires. J. Hazard. Mater. 2007, 144, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhao, X.; Zhang, L.; Zhang, P.; Sheng, L. A theoretical kinetics study on low-temperature reactions of methyl acetate radicals with molecular oxygen. Combust. Flame 2018, 196, 45–53. [Google Scholar] [CrossRef]
- Zhou, C.; Sendt, K.; Haynes, B.S. Experimental and kinetic modelling study of H2S oxidation. Proc. Combust. Inst. 2013, 34, 625–632. [Google Scholar] [CrossRef]
- Oran, E.S.; Gamezo, V.N. Origins of the deflagration-to-detonation transition in gas-phase combustion. Combust. Flame 2007, 148, 4–47. [Google Scholar] [CrossRef]
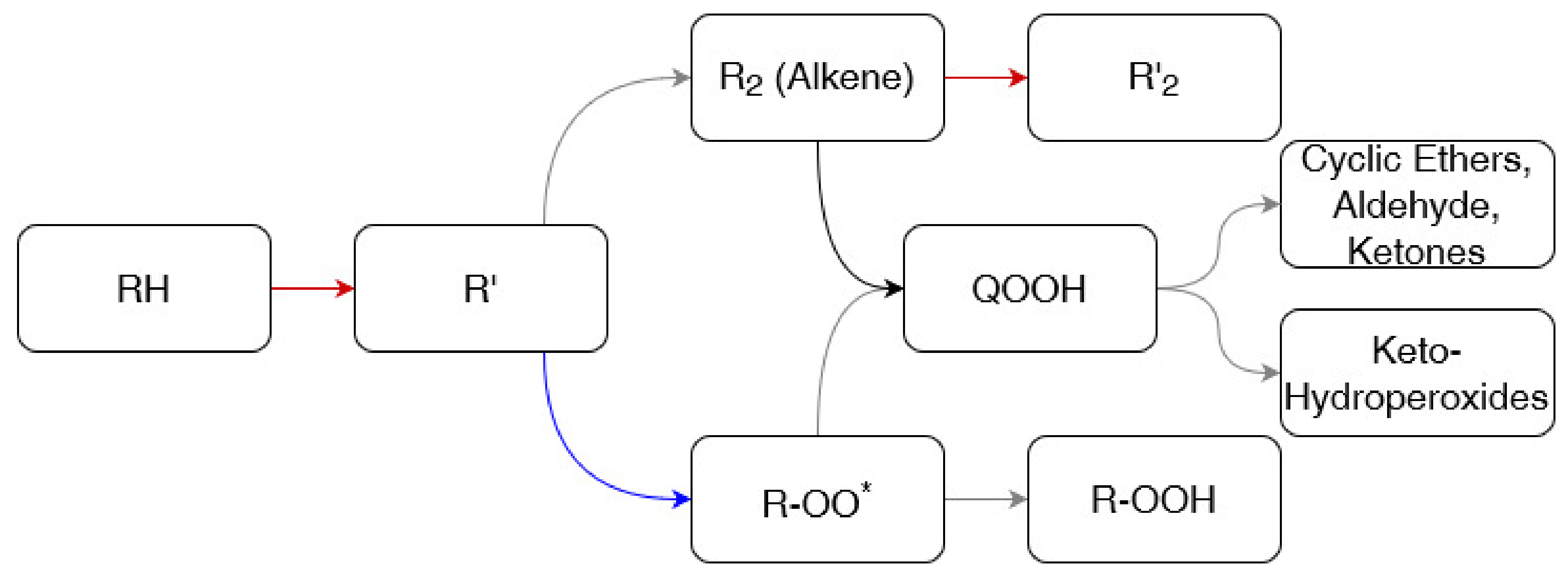
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Walker, R.W.; Morley, C. Chapter 1 Basic chemistry of combustion. Compr. Chem. Kinet. 1997, 35, 1–124. [Google Scholar]
- DeSain, J.D.; Klippenstein, S.J.; Miller, J.A.; Taatjes, C.A. Measurements, theory, and modeling of OH formation in ethyl + O2 and propyl + O2 reactions. J. Phys. Chem. A 2003, 107, 4415–4427. [Google Scholar] [CrossRef]
- Zhang, P.; Ji, W.; He, T.; He, X.; Wang, Z.; Yang, B.; Law, C.K. First-stage ignition delay in the negative temperature coefficient behavior: Experiment and simulation. Combust. Flame 2016, 167, 14–23. [Google Scholar] [CrossRef]
- Curran, H.J.; Gaffuri, P.; Pitz, W.J.; Westbrook, C.K. A comprehensive modeling study of n-heptane oxidation. Combust. Flame 1998, 114, 149–177. [Google Scholar] [CrossRef]
- Battin-Leclerc, F. Detailed chemical kinetic models for the low-temperature combustion of hydrocarbons with application to gasoline and diesel fuel surrogates. Prog. Energy Combust. Sci. 2008, 34, 440–498. [Google Scholar] [CrossRef] [Green Version]
- Warnatz, J. The structure of laminar alkane-, alkene-, and acetylene flames. Symp. Combust. 1981, 18, 369–384. [Google Scholar] [CrossRef]
- Leachman, J.W.; Jacobsen, R.T.; Penoncello, S.G.; Lemmon, E.W. Fundamental equations of state for parahydrogen, normal hydrogen, and orthohydrogen. J. Phys. Chem. Ref. Data 2009, 38, 721–748. [Google Scholar] [CrossRef]
- Navon, N.; Nascimbene, S.; Chevy, F.; Salomon, C. Salomon The Equation of State of a Low-Temperature Fermi Gas with Tunable Interactions. Science 2010, 328, 729–732. [Google Scholar] [CrossRef] [Green Version]
- Tkaczuk, J.; Bell, I.H.; Lemmon, E.W.; Luchier, N.; Millet, F. Equations of State for the Thermodynamic Properties of Binary Mixtures for Helium-4, Neon, and Argon. J. Phys. Chem. Ref. Data 2020, 49, 23101. [Google Scholar] [CrossRef]
- Sakoda, N.; Shindo, K.; Shinzato, K.; Kohno, M.; Takata, Y.; Fujii, M. Review of the Thermodynamic Properties of Hydrogen Based on Existing Equations of State. Int. J. Thermophys. 2010, 31, 276–296. [Google Scholar] [CrossRef]
- Bai-gang, S.; Dong-sheng, Z.; Fu-shui, L. A new equation of state for hydrogen gas. Int. J. Hydrogen Energy 2012, 37, 932–935. [Google Scholar] [CrossRef]
- Nasrifar, K. Comparative study of eleven equations of state in predicting the thermodynamic properties of hydrogen. Int. J. Hydrogen Energy 2010, 35, 3802–3811. [Google Scholar] [CrossRef]
- Hansen, O.R. Liquid hydrogen releases show dense gas behavior. Int. J. Hydrogen Energy 2019, 45, 1343–1358. [Google Scholar] [CrossRef]
- Bubbico, R.; Salzano, E. Acoustic analysis of blast waves produced by rapid phase transition of LNG released on water. Saf. Sci. 2009, 47, 515–521. [Google Scholar] [CrossRef]
- Cleaver, P.; Johnson, M.; Ho, B. Rapid Phase Transition of LNG. J. Hazard. Mater. 2007, 140, 429–438. [Google Scholar] [CrossRef]
- Véchot, L.; Olewski, T.; Osorio, C.; Basha, O.; Liu, Y.; Mannan, S. Laboratory scale analysis of the influence of different heat transfer mechanisms on liquid nitrogen vaporization rate. J. Loss Prev. Process Ind. 2013, 26, 398–409. [Google Scholar] [CrossRef]
- Conrado, C.; Vesovic, V. The influence of chemical composition on vaporisation of LNG and LPG on unconfined water surfaces. Chem. Eng. Sci. 2000, 55, 4549–4562. [Google Scholar] [CrossRef]
- Carslow, H.S.; Jaeger, J.C.; Morral, J.E. Conduction of Heat in Solids, 2nd ed.; Oxford University Press: Oxford, UK, 1986. [Google Scholar] [CrossRef]
- Incropera, F.P.; Dewitt, D.P.; Bergamm, T.L.; Lavine, A.S. Fundamentals of Heat and Mass Transfer, 6th ed.; John Wiley and Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Bird, R.B.; Stewart, W.E.; Lightfoot, E.N. Transport Phenomena, 2nd ed.; Bird, R.B., Stewart, W.E., Lightfoot, E.N., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2002; ISBN 0471410772. [Google Scholar] [CrossRef]
- Persad, A.H.; Ward, C.A. Expressions for the Evaporation and Condensation Coefficients in the Hertz-Knudsen Relation. Chem. Rev. 2016, 116, 7727–7767. [Google Scholar] [CrossRef]
- Nawaz, W.; Olewski, T.; Véchot, L. Assessment and Validation of Evaporation Models for Cryogenic Liquids. Process Saf. Environ. Prot. 2019, 121, 50–61. [Google Scholar] [CrossRef]
- Hertz, H. Ueber die Berührung fester elastischer Körper. J. fur die Reine und Angew. Math. 1882. [Google Scholar] [CrossRef]
- Knudsen, M. Die maximale Verdampfungsgeschwindigkeit des Quecksilbers. Ann. Phys. 1915, 352, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Ingersoll, A.P. Mars: Occurrence of liquid water. Science 1970. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.; Matsugu, R.S. Evaporation rates of liquid hydrocarbon spills on land and water. Can. J. Chem. Eng. 1973, 51, 434–439. [Google Scholar] [CrossRef]
- Lee, W.H. Pressure Iteration Scheme for Two-Phase Flow Modeling, 1st ed.; Hemisphere Publishing: Washington, DC, USA, 1980. [Google Scholar]
- Raj, P.P.K.; Morris, J.A. Source Characterization of Heavy-Gas Dispersion Models for Reactive Chemicals; Technology and Management Systems, Inc.: Burlington, VT, USA, 1987. [Google Scholar]
- Reed, M. The physical fates component of the natural resource damage assessment model system. Oil Chem. Pollut. 1989, 5, 99–123. [Google Scholar] [CrossRef]
- Hummel, A.A.; Braun, K.O.; Fehrenbacher, M.C. Evaporation of a liquid in a flowing airstream. Am. Ind. Hyg. Assoc. J. 1996, 57, 519–525. [Google Scholar] [CrossRef]
- Barry, J. Estimating rates of spreading and evaporation of volatile liquids. Chem. Eng. Prog. 2005, 1, 32–39. [Google Scholar]
- Sears, D.W.G.; Moore, S.R. On laboratory simulation and the evaporation rate of water on Mars. Geophys. Res. Lett. 2005. [Google Scholar] [CrossRef] [Green Version]
- Heymes, F.; Aprin, L.; Bony, A.; Forestier, S.; Cirocchi, S.; Dusserre, G. An experimental investigation of evaporation rates for different volatile organic compounds. Process Saf. Prog. 2013, 32, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schrage, R.W. A Theoretical Study of Interphase Mass Transfer, 1st ed.; Columbia University Press: New York, NY, USA, 1953. [Google Scholar]
- Hissong, D.W. Keys to modeling LNG spills on water. J. Hazard. Mater. 2007, 140, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Morse, T.L.; Kytömaa, H.K. The effect of turbulence on the rate of evaporation of LNG on water. J. Loss Prev. Process Ind. 2011, 24, 791–797. [Google Scholar] [CrossRef]
- Vesovic, V. The influence of ice formation on vaporization of LNG on water surfaces. J. Hazard. Mater. 2007, 140, 518–526. [Google Scholar] [CrossRef]
- Klimenko, V.V. Film boiling on a horizontal plate—New correlation. Int. J. Heat Mass Transf. 1981, 24, 69–79. [Google Scholar] [CrossRef]
- Vílchez, J.A.; Villafane, D.; Casal, J. A dispersion safety factor for LNG vapor clouds. J. Hazard. Mater. 2013, 247, 181–188. [Google Scholar] [CrossRef]
- Rauchegger, C.; Bayley, S.; Schröder, V.; Thévenin, D. Dispersion of heavy gases—Experimental results and numerical simulations. Process Saf. Prog. 2015, 34, 280–285. [Google Scholar] [CrossRef]
- Bosen, J.F. An Approximation Formula to Compute Relative Humidity From Dry Bulb and Dew Point Temperatures. Mon. Weather Rev. 1958. [Google Scholar] [CrossRef]
- Àgueda, A.; Subirana, J.; Pastor, E.; Schleder, A.M.; Planas, E. Revisiting the dispersion safety factor (DSF) for vapor clouds of liquefied flammable gases (LNG and propane). Saf. Sci. 2020, 128, 104748. [Google Scholar] [CrossRef]
- Aursand, E.; Hammer, M. Predicting triggering and consequence of delayed LNG RPT. J. Loss Prev. Process Ind. 2018, 55, 124–133. [Google Scholar] [CrossRef]
- Spiegler, P.; Hopenfeld, J.; Silberberg, M.; Bumpus, C.F.; Norman, A. Onset of stable film boiling and the foam limit. Int. J. Heat Mass Transf. 1963, 6, 987–989. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Zhang, F.; Xie, F.; Ma, Y. Correlations for calculating heat transfer of hydrogen pool boiling. Int. J. Hydrogen Energy 2016, 41, 17118–17131. [Google Scholar] [CrossRef]
- Enger, T.; Hartman, D.E. LNG Spillage on Water. II. Final Report on Rapid Phase Transitions; Shell Pipeline Corp., Research and Development Laboratory: Houston, TX, USA, 1972. [Google Scholar]
- Ermak, D.L.; Koopman, R.P.; McRae, T.G.; Hogan, W.J. Lng spill experiments: Dispersion, RPT, and vapor burn analysis. In Proceedings of the American Gas Association, Operating Section, Washington, DC, USA, 3–5 May 1982; p. 32. [Google Scholar]
- Luketa-Hanlin, A. A review of large-scale LNG spills: Experiments and modeling. J. Hazard. Mater. 2006. [Google Scholar] [CrossRef] [PubMed]
- Pitblado, R.M.; Woodward, J.L. Highlights of LNG risk technology. J. Loss Prev. Process Ind. 2011, 24, 827–836. [Google Scholar] [CrossRef]
- Drake, E.M.; Jeje, A.A.; Reid, R.C. Transient boiling of liquefied cryogens on a water surface. II. Light hydrocarbon mixtures. Int. J. Heat Mass Transf. 1975, 18, 1369–1375. [Google Scholar] [CrossRef]
- Porteous, W.; Blander, M. Limits of superheat and explosive boiling of light hydrocarbons, halocarbons, and hydrocarbon mixtures. AIChE J. 1975, 21, 560–566. [Google Scholar] [CrossRef]
- Blander, M.; Katz, J.L. Bubble nucleation in liquids. AIChE J. 1975, 21, 833–848. [Google Scholar] [CrossRef]
- Feldbauer, G.F.; Heigl, J.J.; McQueen, W.; Whipp, R.H.; May, W.G. Spills of LNG on Water-Vaporization and Downwind Drift of Combustible Mixtures; ESSO Research and Engineering Co.: Florham Park, NJ, USA, 1972. [Google Scholar]
- Puttock, J.S.; Blackmore, D.R.; Colenbrander, G.W. Field experiments on dense gas dispersion. J. Hazard. Mater. 1982, 6, 13–41. [Google Scholar] [CrossRef]
- Koopman, R.P.; Cederwal, L.R.T.; Ermak, E.R.; Goldwire, H.C.; Hogan, W.J.; McClure, J.W.; McRae, T.G.; Morgan, D.L.; Rodean, H.C.; Shinn, J.H. Burro Series Data Report LLNL/NWC 1980 LNG Spill Tests, UCID-19075; Lawrence Livermore National Laboratory: Berkeley, CA, USA, 1980.
- Brown, T.C.; Cederwall, R.T.; Chan, S.T.; Ermak, D.L.; Koopman, R.P.; Lamson, K.C.; McClure, J.W.; Morris, L.K. Falcon Series Data Report: 1987 LNG Vapor Barrier Verification Field Trials; P&N Publications: Bannockburn, UK, 1987. [Google Scholar]
- Cormier, B.R.; Qi, R.; Yun, G.W.; Zhang, Y.; Sam Mannan, M. Application of computational fluid dynamics for LNG vapor dispersion modeling: A study of key parameters. J. Loss Prev. Process Ind. 2009, 22, 332–352. [Google Scholar] [CrossRef]
- Lu, T.; Law, C.K. Toward accommodating realistic fuel chemistry in large-scale computations. Prog. Energy Combust. Sci. 2009, 35, 192–215. [Google Scholar] [CrossRef]
- Varma, A.; Morbidelli, M.; Wu, H. Parametric Sensitivity in Chemical Systems, 1st ed.; Cambridge University Press: Cambridge, UK, 1999; ISBN 13-978-0-521-62171-7. [Google Scholar] [CrossRef]
- Ouyang, L.; Li, H.; Sun, S.; Wang, X.; Lu, X. Auto-ignition of biomass synthesis gas in shock tube at elevated temperature and pressure. Sci. Bull. 2015, 60, 1935–1946. [Google Scholar] [CrossRef]
- Rengel, B.; Mata, C.; Pastor, E.; Casal, J.; Planas, E. A priori validation of CFD modelling of hydrocarbon pool fires. J. Loss Prev. Process Ind. 2018, 56, 18–31. [Google Scholar] [CrossRef]
- Van den Bosch, C.; Weterings, R. Yellow Book—Methods for the Calculation of Physical Effects Due to Releases of Hazardous Materials (Liquids and Gases), 3rd ed.; TNO: Hague, The Netherlands, 1997. [Google Scholar]
- Mcgrattan, K.B.; Baum, H.R.; Hamins, A. Thermal Radiation from Large Pool Fires; NIST: Gaithersburg, MD, USA, 2000.
- Beyler, C.L. Industrial Fire Protection Engineering, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; Volume 40, ISBN 0471496774. [Google Scholar] [CrossRef]
- Babrauskas, V. Estimating large pool fire burning rates. Fire Technol. 1983, 19, 251–261. [Google Scholar] [CrossRef]
- Fay, J.A. Model of large pool fires. J. Hazard. Mater. 2006, 136, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.H. The size of flames from natural fires. Symp. Combust. 1963, 9, 844–859. [Google Scholar] [CrossRef]
- Phillips, H. Flame in a buoyant methane layer. In Proceedings of the Symposium (International) on Combustion, Pittsburgh, PA, USA, 1 January 1965. [Google Scholar] [CrossRef]
- Pio, G.; Salzano, E. Laminar Burning Velocity of Methane, Hydrogen, and Their Mixtures at Extremely Low-Temperature Conditions. Energy Fuels 2018, 32, 8830–8836. [Google Scholar] [CrossRef]
- Liang, T.; Zhong, W.; Yuen, R.K.K.; Lo, S.; Liao, G. On the fire intensification of pool fire with water mist. Procedia Eng. 2013, 62, 994–999. [Google Scholar] [CrossRef] [Green Version]
- Rana, M.A.; Guo, Y.; Mannan, M.S. Use of water spray curtain to disperse LNG vapor clouds. J. Loss Prev. Process Ind. 2010, 23, 77–88. [Google Scholar] [CrossRef]
- Suardin, J.A.; Qi, R.; Cormier, B.R.; Rana, M.; Zhang, Y.; Mannan, M.S. Application of fire suppression materials on suppression of LNG pool fires. J. Loss Prev. Process Ind. 2011, 24, 63–75. [Google Scholar] [CrossRef]
- Raj, P.K. Large LNG fire thermal radiation—Modeling issues and hazard criteria revisited. Process Saf. Prog. 2005. [Google Scholar] [CrossRef]
- Colenbrander, G.W.; Puttock, J.S. Maplin Sands experiments 1980: Interpretation and modelling of liquefied gas spills onto the sea. In Atmospheric Dispersion of Heavy Gases and Small Particles; Springer: Berlin/Heidelberg, Germany, 1984; pp. 277–295. [Google Scholar] [CrossRef]
- Malvos, H.; Raj, P. Details of 35 m diameter LNG fire tests conducted in Montoir, France in 1987, and analysis of fire spectral and other data. In Proceedings of the 2006 AIChE Spring Annual Meeting, Orlando, FL, USA, 23–27 April 2006. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | A0 | A1 | A2 | A3 | A4 | A5 | A6 |
|---|---|---|---|---|---|---|---|
| n-H2 | 3.50 × 100 | 7.97 × 10−5 | −2.55 × 10−7 | 3.61 × 10−11 | 2.69 × 10−13 | −1.06 × 103 | −4.28 × 100 |
| o-H2 | 2.55 × 100 | −1.96 × 10−3 | 4.08 × 10−6 | 2.06 × 10−7 | −5.69 × 10−10 | −9.04 × 102 | 1.81 × 100 |
| p-H2 | 2.61 × 100 | −1.39 × 10−2 | 3.39 × 10−4 | −1.61 × 10−6 | 2.22 × 10−9 | −1.06 × 103 | −3.59 × 10−1 |
| CH4 | 4.21 × 100 | −5.36 × 10−3 | 2.51 × 10−5 | −2.14 × 10−8 | 5.97 × 10−12 | −1.02 × 104 | −9.21 × 10−1 |
| C2H6 | 3.75 × 100 | 4.55 × 10−5 | 4.08 × 10−5 | −4.57 × 10−8 | 1.57 × 10−11 | −1.15 × 104 | 4.74 × 100 |
| C3H8 | 3.06 × 100 | 1.29 × 10−2 | 3.47 × 10−5 | −4.71 × 10−8 | 1.71 × 10−11 | −1.44 × 104 | 1.08 × 101 |
| N2 | 3.54 × 100 | −6.93 × 10−4 | 2.10 × 10−6 | −1.29 × 10−9 | 2.59 × 10−13 | −1.04 × 103 | 2.99 × 100 |
| O2 | 3.12 × 100 | 1.73 × 10−3 | −8.53 × 10−7 | 1.70 × 10−10 | −1.23 × 10−14 | −1.04 × 103 | 6.28 × 100 |
| Characteristic | Typical Alternatives |
|---|---|
| Explosion vessel | Closed glass sphere or cylinder, Steel tube, Glass flask, Quartz tube |
| Flame direction | Horizontal, Vertical |
| Ignition source | Spark, Flame, Exploding wire |
| Ignition duration | 0.2 s, 0.5 s |
| Ignition position | Sphere centre, Tube bottom |
| Ignition criteria | Visual flame, Thermal rise, Pressure rise, Pressure-time curve inflexion point |
| Limit definition | Last non-ignition point, Average of last ignition and non-ignition points |
| Mixture | CH4 [% vol/vol] | C2H6 [%vol/vol] | C3H8 [%vol/vol] |
|---|---|---|---|
| LNG 1 | 100 | 0 | 0 |
| LNG 2 | 90 | 10 | 0 |
| LNG 3 | 80 | 10 | 10 |
| Authors [Reference] | Equation |
|---|---|
| Hertz-Knudsen-Schrage [115,116] | |
| Ingersoll [117] | |
| Mackay and Matsugu [118] | |
| Lee [119] | |
| Raj and Morris [120] | |
| Reed [121] | |
| Hummel [122] | |
| Barry [123] | |
| Derek et al. [124] | |
| Heymes [125] | |
| Schrage [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pio, G.; Salzano, E. Accidental Combustion Phenomena at Cryogenic Conditions. Safety 2021, 7, 67. https://doi.org/10.3390/safety7040067
Pio G, Salzano E. Accidental Combustion Phenomena at Cryogenic Conditions. Safety. 2021; 7(4):67. https://doi.org/10.3390/safety7040067
Chicago/Turabian StylePio, Gianmaria, and Ernesto Salzano. 2021. "Accidental Combustion Phenomena at Cryogenic Conditions" Safety 7, no. 4: 67. https://doi.org/10.3390/safety7040067
APA StylePio, G., & Salzano, E. (2021). Accidental Combustion Phenomena at Cryogenic Conditions. Safety, 7(4), 67. https://doi.org/10.3390/safety7040067