Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma



Abstract
:
1. Introduction
- Poor cell internalization. Their negative charge prevents passive endocytosis [17].
- Poor cellular selectivity. The oligonucleotides cannot discriminate between healthy and cancer cells, leading to a non-specific delivery [37].
2. Materials and Methods
2.1. Materials
2.2. Modified Oligonucleotides
2.3. Oligonucleotide Solution Preparation
2.3.1. RNA Annealing (siRNAs)
2.3.2. siRNA Mix
2.3.3. DNA Mix
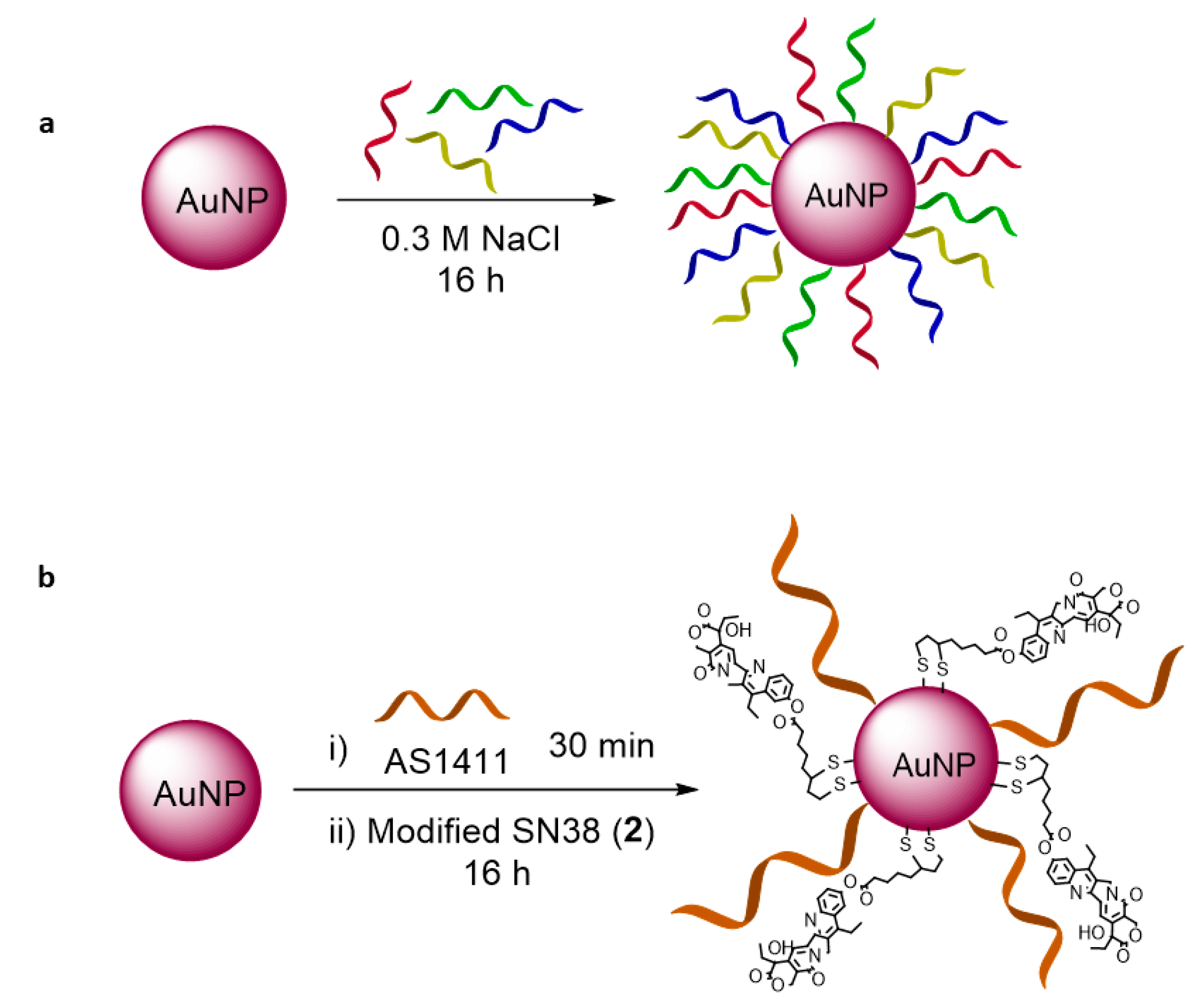
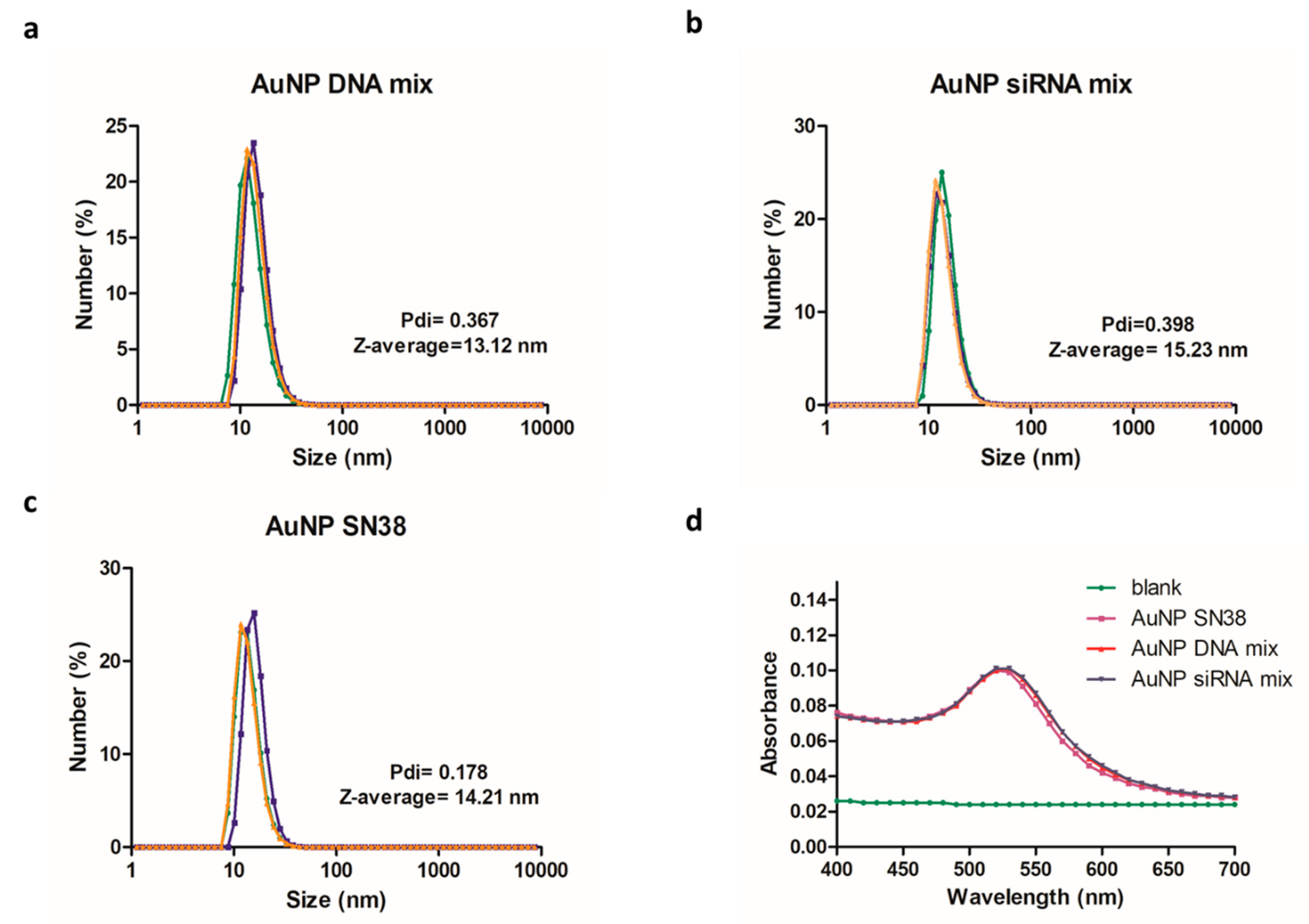
2.4. Gold Nanoparticles Synthesis and Characterization
Gold Nanoparticle Functionalization
2.5. Cell Culture and Viability Studies
2.5.1. alamarBlue Viability Assay
2.5.2. Oligonucleotide Transfection
2.5.3. Chemotherapy
2.5.4. Combination Treatment
2.5.5. Nanoparticles Treatment
2.6. c-Met Immunofluorescence
2.7. Flow Cytometry
2.8. Synthesis of Modified SN38
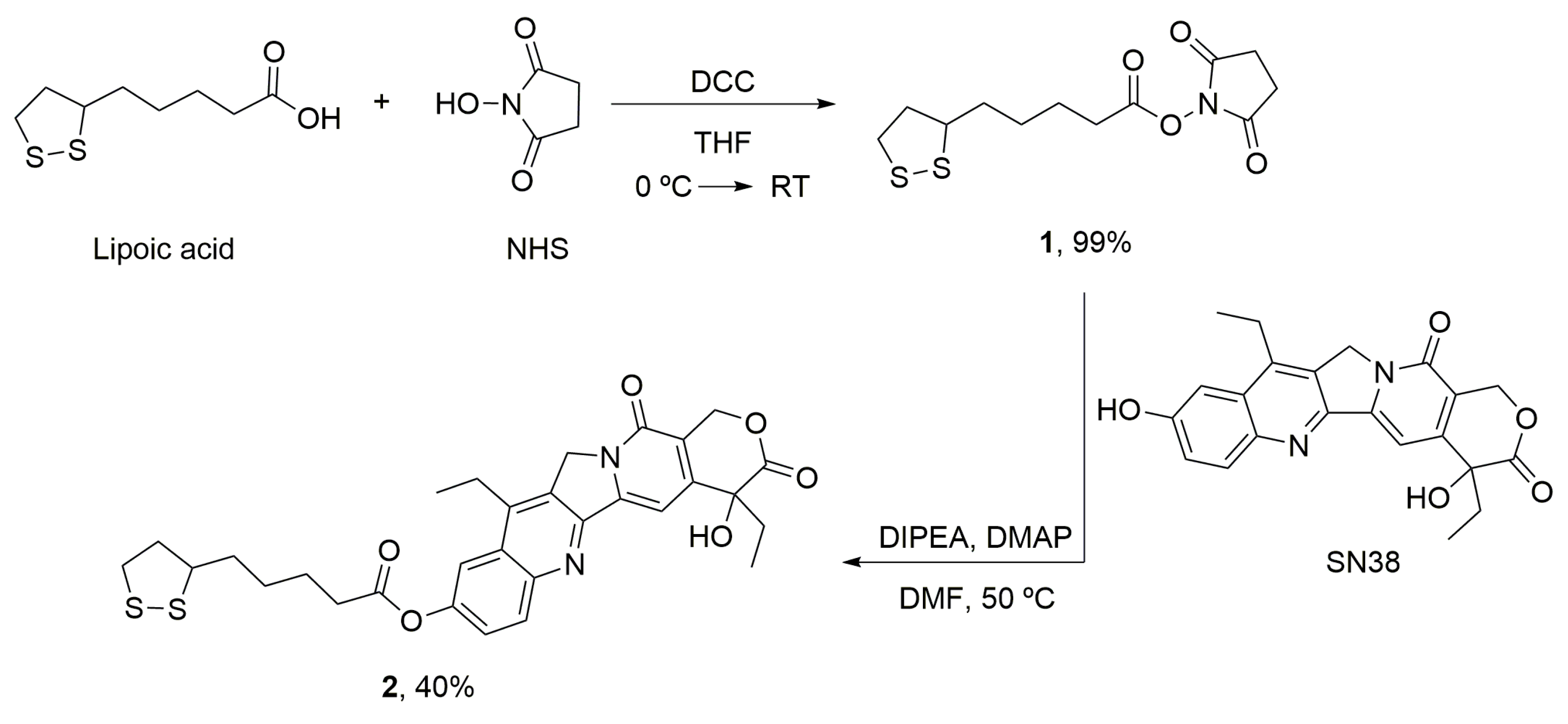
2.8.1. α-Lipoic Acid–NHS (1)
2.8.2. α-Lipoic Acid–SN38 (2)
2.9. Statistical Analysis
3. Results
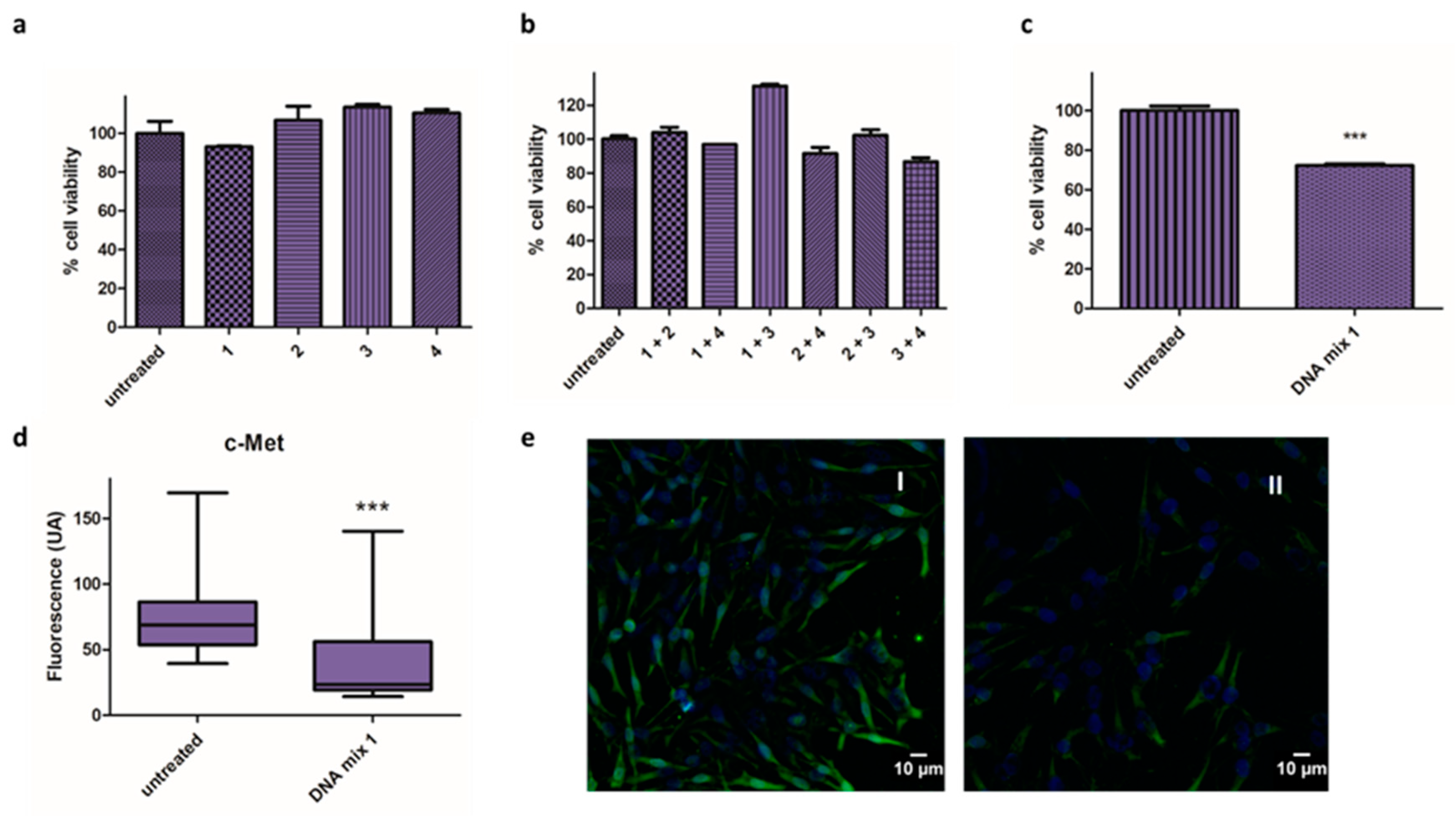
3.1. DNA-Based microRNA Mimics
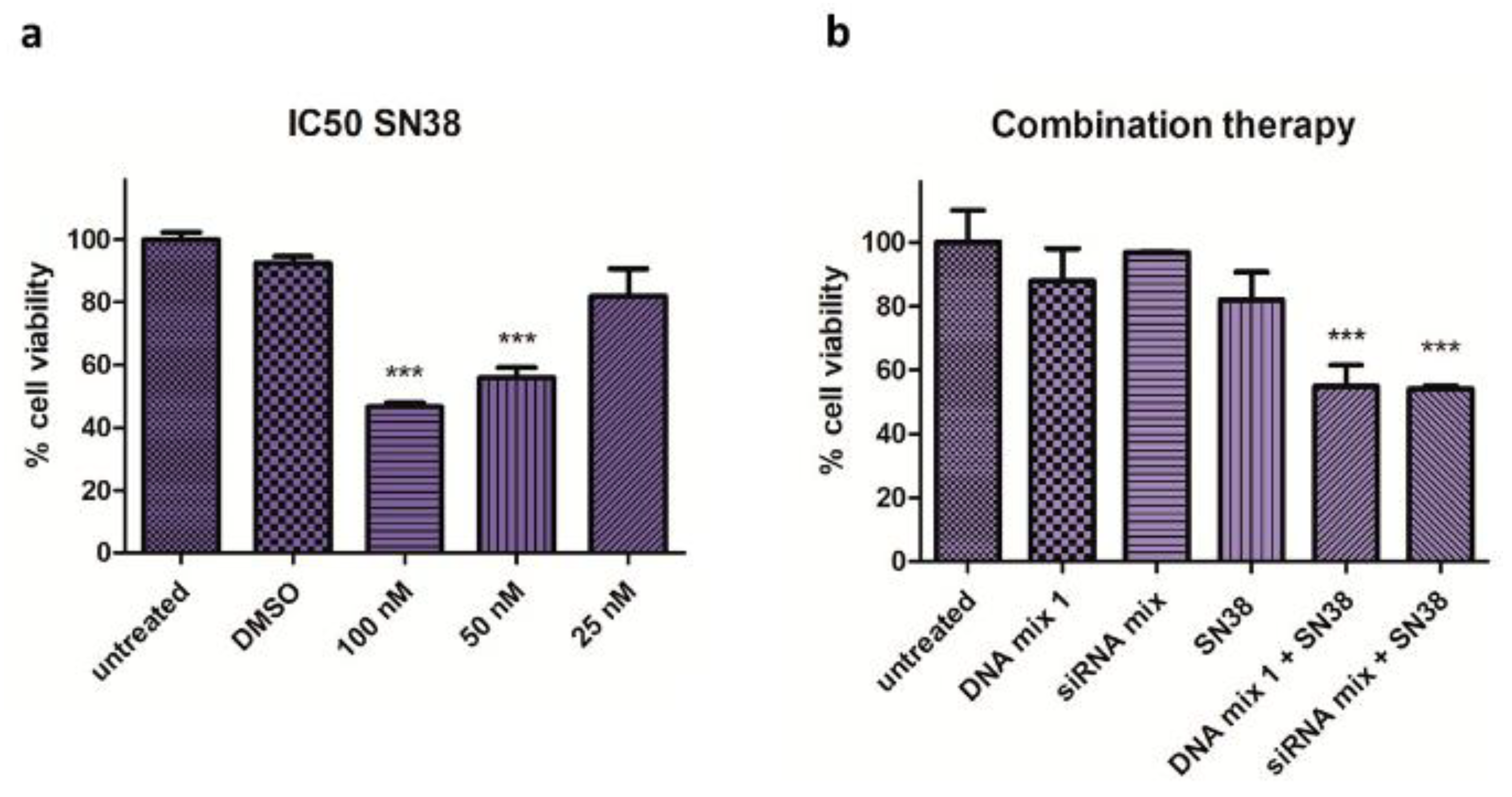
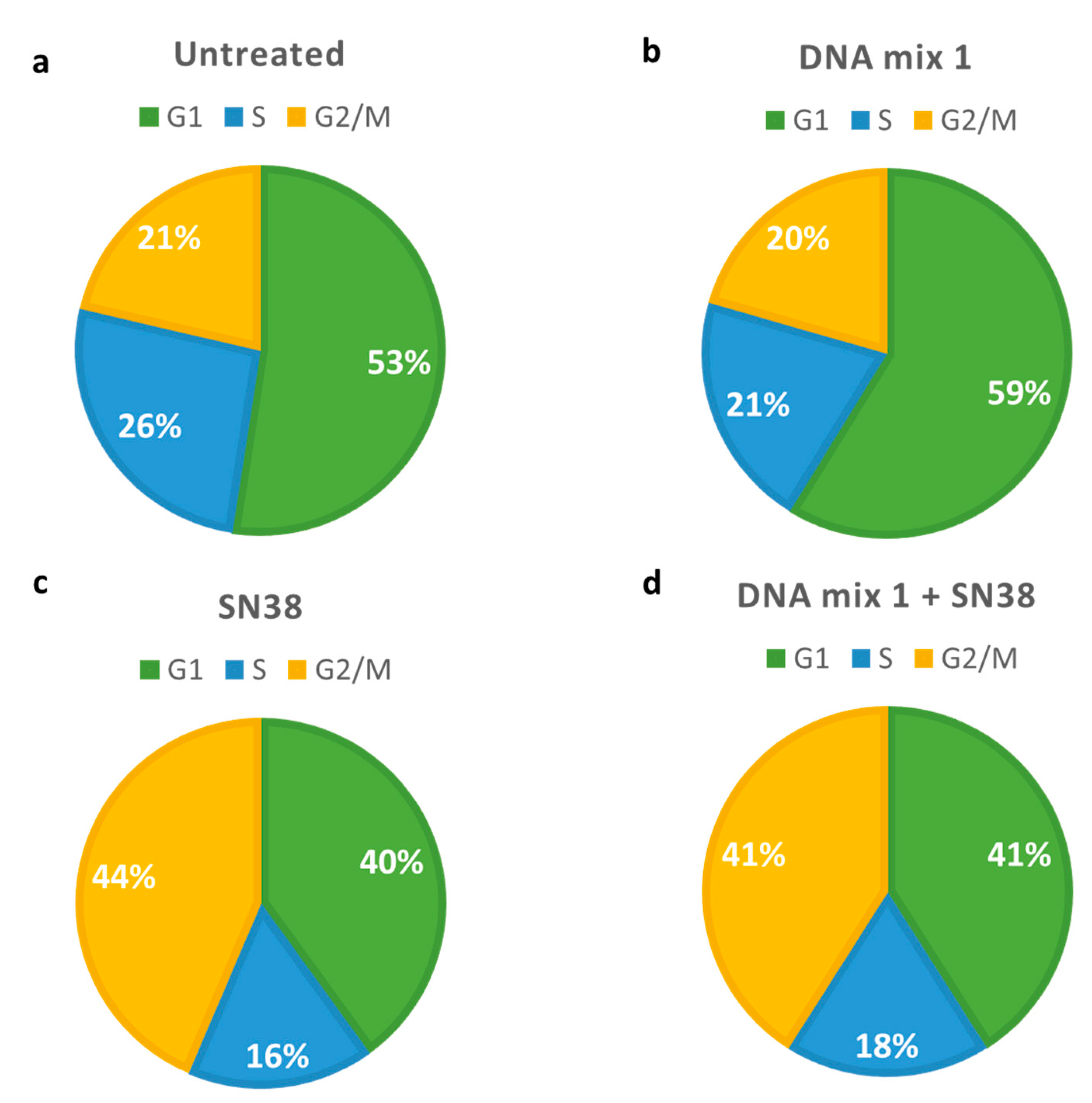
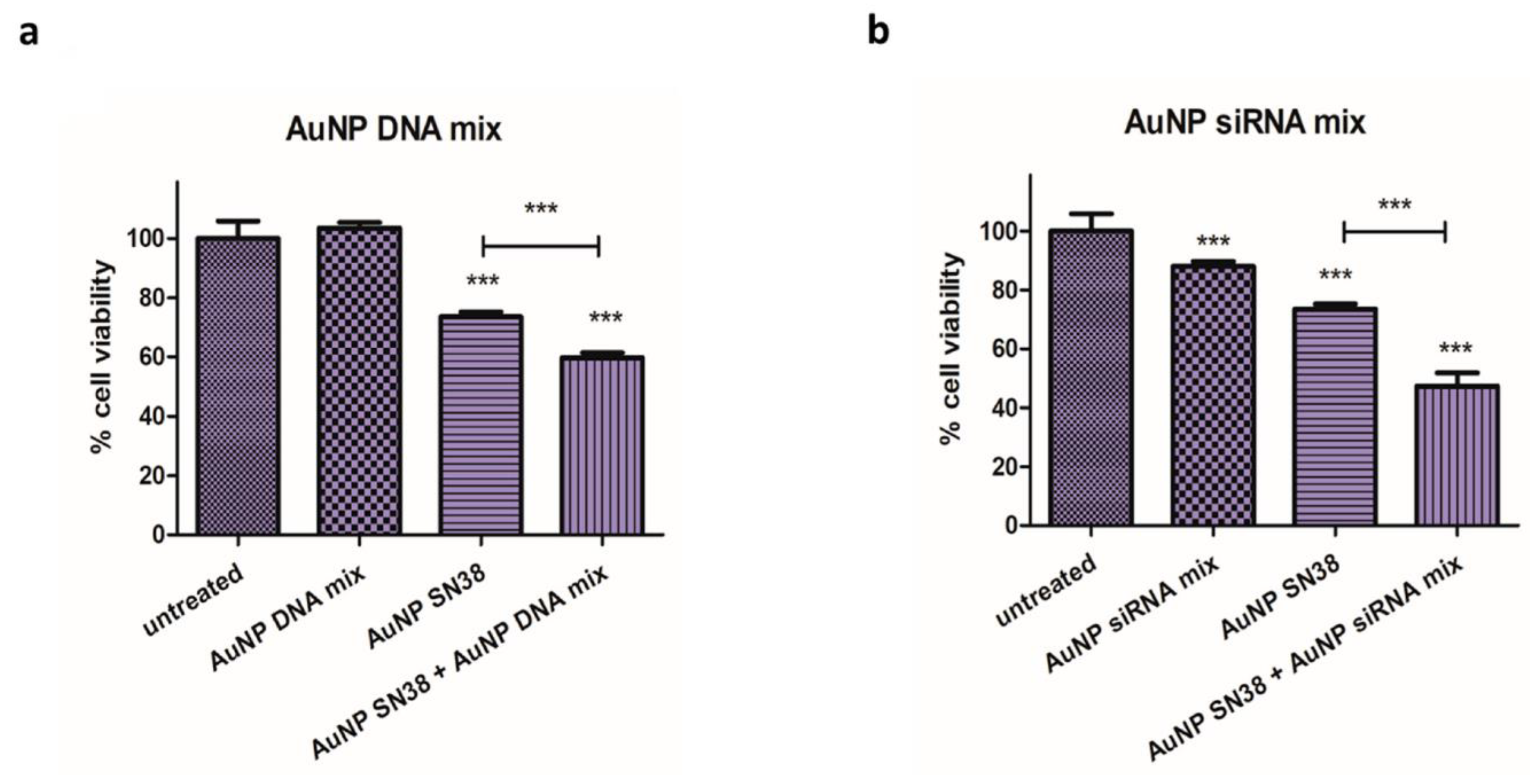
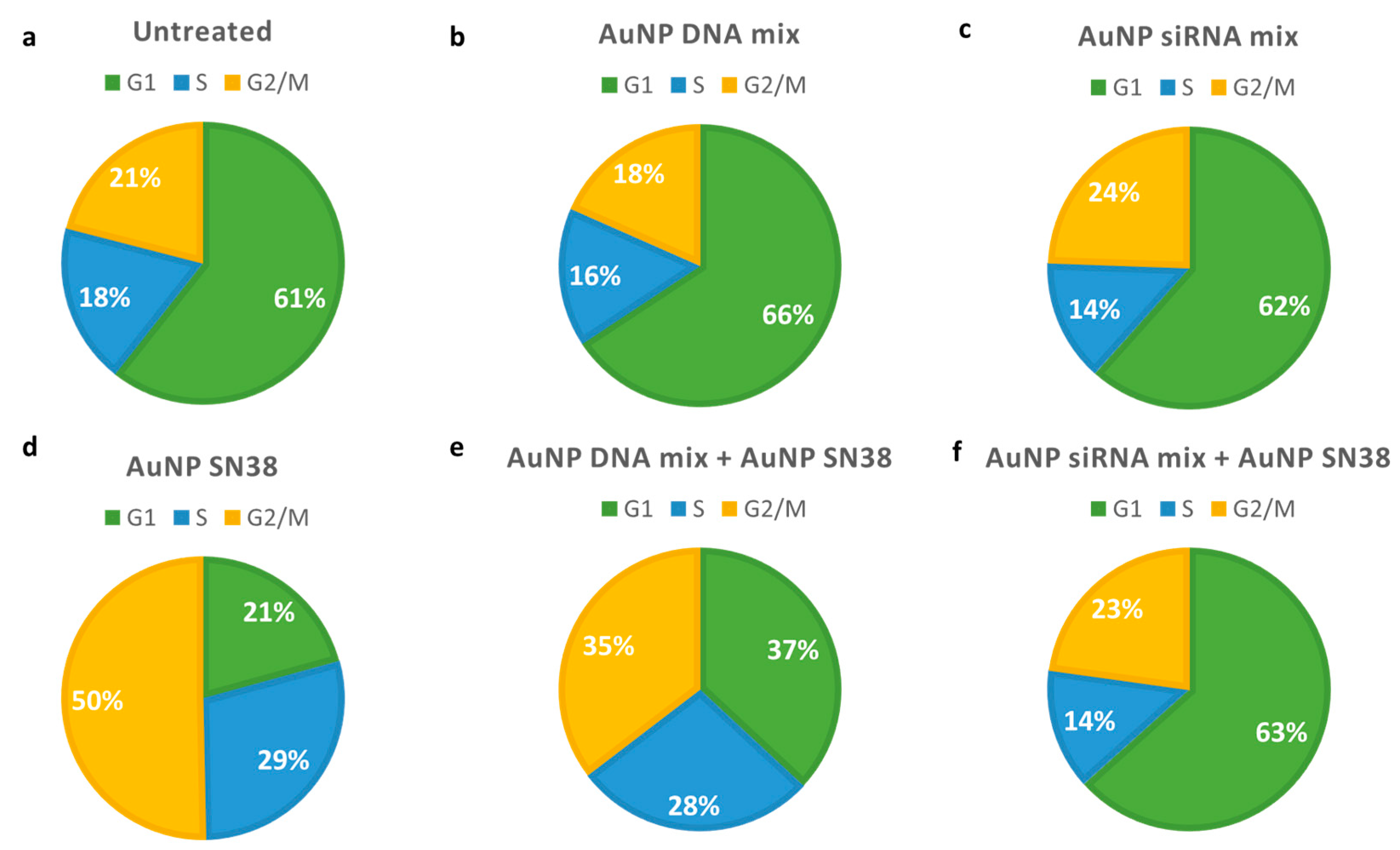
3.2. Combination Therapy
3.3. Gold Nanoparticles as Carriers
4. Discussion
4.1. DNA-Based microRNA Mimics
4.2. Combination Therapy
4.3. Gold Nanoparticles as Vehicles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Corrêa, Z.M. Assessing prognosis in uveal melanoma. Cancer Control 2016, 23, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Rodríguez, B.; Latorre, A.; Posch, C.; Somoza, Á. Recent advances in uveal melanoma treatment. Med. Res. Rev. 2017, 37, 1350–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichstein, D. New concepts in the molecular understanding of uveal melanoma. Curr. Opin. Ophthalmol. 2017, 28, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Terheyden, P.; Kämpgen, E.; Wagner, S.; Neumann, C.; Schadendorf, D.; Steinmann, A.; Wittenberg, G.; Lieb, W.; Bröcker, E.-B. Treatment of disseminated ocular melanoma with sequential fotemustine, interferon α and interleukin 2. Br. J. Cancer 2002, 87, 840–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentini, G.; Aliberti, C.; Benea, G.; Tilli, M.; Del Conte, A. A pilot study of trans arterial chemo-embolization (TACE) with drug eluting beads irinotecan (IRI) preloaded (DEBI) for liver metastases (LM) from uveal melanoma (UM): An Italian National Study. J. Clin. Oncol. 2008, 26, 20010. [Google Scholar] [CrossRef]
- Carling, U.; Dorenberg, E.J.; Haugvik, S.-P.; Eide, N.A.; Berntzen, D.T.; Edwin, B.; Dueland, S.; Røsok, B. Transarterial chemoembolization of liver metastases from uveal melanoma using irinotecan-loaded beads: Treatment response and complications. Cardiovasc. Interv. Radiol. 2015, 38, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Martens-Uzunova, E.S.; Olvedy, M.; Jenster, G. Beyond microRNA—Novel RNAs derived from small non-coding RNA and their implication in cancer. Cancer Lett. 2013, 340, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Girnita, L.; Buda, O.; Calin, G.A. Non-coding RNAs: The cancer genome dark matter that matters! Clin. Chem. Lab. Med. 2017, 55, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, M.; Tuccoli, A.; Poliseno, L. Long non-coding RNAs in cancer: Implications for personalized therapy. Cell. Oncol. 2015, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, C.B.; Calcagno, D.Q.; Araújo, T.M.T.; Batista dos Santos, S.E.; Ribeiro dos Santos, Â.K.C.; Riggins, G.J.; Burbano, R.R.; Assumpção, P.P. The role of piRNA and its potential clinical implications in cancer. Epigenomics 2015, 7, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Xu, T.; Ganapathy, S.; Shadfan, M.; Long, M.; Huang, T.H.-M.; Thompson, I.; Yuan, Z.-M. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 2014, 33, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Barciszewska, A.-M. MicroRNAs as efficient biomarkers in high-grade gliomas. Folia Neuropathol. 2016, 4, 369–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yu, X.; Shen, J.; Jiang, Y. MicroRNA dysregulation in uveal melanoma: A new player enters the game. Oncotarget 2015, 6, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Chen, Y.; Leong, K.W. MicroRNA delivery for regenerative medicine. Adv. Drug Deliv. Rev. 2015, 88, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekris, L.M.; Leverenz, J.B. The biomarker and therapeutic potential of miRNA in Alzheimer’s disease. Neurodegener. Dis. Manag. 2015, 5, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhou, W.; Li, C.; Guo, M. MicroRNAs, DNA damage response, and cancer treatment. Int. J. Mol. Sci. 2016, 17, 2087–2101. [Google Scholar] [CrossRef] [PubMed]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell. Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Invest. 2017, 127, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Sun, B.; Su, C. Targeting microRNAs in cancer gene therapy. Genes (Basel) 2017, 8, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, X.; Shen, J.; Liu, Y.; Chan, M.T.V.; Wu, W.K.K. MicroRNA dysregulation in rhabdomyosarcoma: A new player enters the game. Cell Prolif. 2015, 48, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Han, F.; Yamamoto, A. The biology and management of uveal melanoma. Curr. Oncol. Rep. 2008, 10, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Tan, C. Combination of microRNA therapeutics with small-molecule anticancer drugs: Mechanism of action and co-delivery nanocarriers. Adv. Drug Deliv. Rev. 2015, 81, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Zhou, X.; Chen, X.; Hu, D.-N.; Dong, X.D.; Wang, J.; Lu, F.; Tu, L.; Qu, J. MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Investig. Opthalmology Vis. Sci. 2009, 50, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Gramantieri, L.; Granito, A.; Guidetti, E. c-MET receptor tyrosine kinase as a molecular target in advanced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2015, 2, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Lei, R.; Hu, G. Roles of miR-182 in sensory organ development and cancer. Thorac. Cancer 2015, 6, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eedunuri, V.K.; Rajapakshe, K.; Fiskus, W.; Geng, C.; Chew, S.A.; Foley, C.; Shah, S.S.; Shou, J.; Mohamed, J.S.; Coarfa, C.; et al. miR-137 targets p160 steroid receptor coactivators SRC1, SRC2, and SRC3 and inhibits cell proliferation. Mol. Endocrinol. 2015, 29, 1170–1183. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y. MicroRNA-137 Targets carboxyl-terminal binding protein 1 in melanoma cell lines. Int. J. Biol. Sci. 2011, 7, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bian, G.; Meng, Z.; Dang, G.; Shi, D.; Mi, S. MiR-144 inhibits uveal melanoma cell proliferation and invasion by regulating c-Met expression. PLoS ONE 2015, 10, e0124428. [Google Scholar] [CrossRef] [PubMed]
- Tutar, L.; Tutar, E.; Tutar, Y. MicroRNAs and cancer; An overview. Curr. Pharm. Biotechnol. 2014, 15, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Mognato, M.; Celotti, L. MicroRNAs used in combination with anti-cancer treatments can enhance therapy efficacy. Mini-Rev. Med. Chem. 2015, 15, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Leonetti, C.; Zupi, G. The future of antisense therapy: Combination with anticancer treatments. Oncogene 2003, 22, 6579–6588. [Google Scholar] [CrossRef] [PubMed]
- Vinhas, R.; Fernandes, A.R.; Baptista, P. V Gold Nanoparticles for BCR-ABL1 gene silencing: Improving tyrosine kinase inhibitor efficacy in chronic myeloid leukemia. Mol. Ther. Nucleic Acids 2017, 7, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Villanueva, D.; El-Sherbiny, I.M.; Herrera-Ruiz, D.; Vlassov, A.V.; Smyth, H.D.C. Formulation approaches to short interfering RNA and microRNA: Challenges and implications. J. Pharm. Sci. 2012, 101, 4046–4066. [Google Scholar] [CrossRef] [PubMed]
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Gou, Y.; Miao, D.; Zhou, M.; Wang, L.; Zhou, H.; Su, G. Bio-inspired protein-based nanoformulations for cancer theranostics. Front. Pharmacol. 2018, 9, 421–462. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Patharkar, A.; Kuche, K.; Maheshwari, R.; Deb, P.K.; Kalia, K.; Tekade, R.K. Functionalized carbon nanotubes as emerging delivery system for the treatment of cancer. Int. J. Pharm. 2018, 548, 540–558. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Pandit, S.; Mokkapati, V.R.S.S.; Garg, A.; Ravikumar, V.; Mijakovic, I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int. J. Mol. Sci. 2018, 19, 1979. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.K.; Shandilya, R.; Mishra, P.K. Lipid based nanocarriers: A translational perspective. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2023–2050. [Google Scholar] [CrossRef] [PubMed]
- Núñez, C.; Estévez, S.V.; del Pilar Chantada, M. Inorganic nanoparticles in diagnosis and treatment of breast cancer. J. Biol. Inorg. Chem. 2018, 23, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Vallet-Regí, M. Mesoporous silica nanoparticles in nanomedicine applications. J. Mater. Sci. Mater. Med. 2018, 29, 65. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New developments in liposomal drug delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.F. Viral nanoparticles in drug delivery and imaging. Mol. Pharm. 2013, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Posch, C.; Garcimartín, Y.; Celli, A.; Sanlorenzo, M.; Vujic, I.; Ma, J.; Zekhtser, M.; Rappersberger, K.; Ortiz-Urda, S.; et al. DNA and aptamer stabilized gold nanoparticles for targeted delivery of anticancer therapeutics. Nanoscale 2014, 6, 7436–7442. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, C.H.; Melamed, J.R.; Day, E.S. Spherical nucleic acid nanoparticles: Therapeutic potential. BioDrugs 2018, 32, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Cutler, J.I.; Auyeung, E.; Mirkin, C.A. Spherical nucleic acids. J. Am. Chem. Soc. 2012, 134, 1376–1391. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Tekade, R.K.; Chougule, M.B. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: Current progress and advances. J. Control. Release 2014, 194, 238–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Zhang, X.; Gu, Z.; Zhao, Y. Recent advances in upconversion nanoparticles-based multifunctional nanocomposites for combined cancer therapy. Adv. Mater. 2015, 27, 7692–7712. [Google Scholar] [CrossRef] [PubMed]
- Palakurthi, S. Challenges in SN38 drug delivery: Current success and future directions. Expert Opin. Drug Deliv. 2015, 12, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Xuan, T.; Parmar, M.; Ma, L.; Ugwu, S.; Ali, S.; Ahmad, I. Development and characterization of a novel liposome-based formulation of SN-38. Int. J. Pharm. 2004, 270, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Rubio, B.; Sapra, P.; Wu, D.; Reddy, P.; Sai, P.; Martinez, A.; Gao, Y.; Lozanguiez, Y.; Longley, C.; et al. Novel prodrugs of SN38 using multiarm poly(ethylene glycol) linkers. Bioconjug. Chem. 2008, 19, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Protocol for Annealing Oligonucleotides. Available online: https://www.sigmaaldrich.com/technical-documents/protocols/biology/annealing-oligos.html (accessed on 4 July 2018).
- Posch, C.; Latorre, A.; Crosby, M.B.; Celli, A.; Latorre, A.; Vujic, I.; Sanlorenzo, M.; Green, G.A.; Weier, J.; Zekhtser, M.; et al. Detection of GNAQ mutations and reduction of cell viability in uveal melanoma cells with functionalized gold nanoparticles. Biomed. Microdevices 2015, 17, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Navarra, G.; Moschetti, M.; Guarrasi, V.; Mangione, M.R.; Militello, V.; Leone, M. Simultaneous determination of caffeine and chlorogenic acids in green coffee by UV/Vis Spectroscopy. J. Chem. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Prigodich, A.E.; Seferos, D.S.; Massich, M.D.; Giljohann, D.A.; Lane, B.C.; Mirkin, C.A. Nano-flares for mRNA regulation and detection. ACS Nano 2009, 3, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Millstone, J.E.; Georganopoulou, D.G.; Xu, X.; Wei, W.; Li, S.; Mirkin, C.A. DNA-gold triangular nanoprism conjugates. Small 2008, 4, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Del Moral, Á. Cell Fluorescence Weber. Available online: https://github.com/DelmoPy/Cell-Fluorescence-Weber (accesed on 5 July 2018).
- Benito-Alifonso, D.; Tremel, S.; Hou, B.; Lockyear, H.; Mantell, J.; Fermin, D.J.; Verkade, P.; Berry, M.; Galan, M.C. Lactose as a “Trojan Horse” for quantum dot cell transport. Angew. Chem. Int. Ed. 2014, 53, 810–814. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. [Google Scholar]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Khan, S.; Ebeling, M.C.; Zaman, M.S.; Sikander, M.; Yallapu, M.M.; Chauhan, N.; Yacoubian, A.M.; Behrman, S.W.; Zafar, N.; Kumar, D.; et al. MicroRNA-145 targets MUC13 and suppresses growth and invasion of pancreatic cancer. Oncotarget 2014, 5, 7599–7609. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.W.; Cho, W.C. The emerging role of miRNAs in combined cancer therapy. Expert Opin. Biol. Ther. 2015, 15, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Dong, X.D.; Chen, X.; Yao, S.; Wang, L.; Wang, J.; Wang, C.; Hu, D.-N.; Qu, J.; Tu, L. Role of microRNA-182 in posterior uveal melanoma: Regulation of tumor development through MITF, BCL2 and cyclin D2. PLoS ONE 2012, 7, e40967. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhou, J.; Rogers, A.M.; Jänne, P.A.; Benedettini, E.; Loda, M.; Hodi, F.S. c-Met, epidermal growth factor receptor, and insulin-like growth factor-1 receptor are important for growth in uveal melanoma and independently contribute to migration and metastatic potential. Melanoma Res. 2012, 22, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Barisione, G.; Fabbi, M.; Gino, A.; Queirolo, P.; Orgiano, L.; Spano, L.; Picasso, V.; Pfeffer, U.; Mosci, C.; Jager, M.J.; et al. Potential role of soluble c-Met as a new candidate biomarker of metastatic uveal melanoma. JAMA Ophthalmol. 2015, 133, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Boominathan, R.; Foulk, B.; Rao, C.; Kemeny, G.; Strickler, J.H.; Abbruzzese, J.L.; Harrison, M.R.; Hsu, D.S.; Healy, P.; et al. Development of a novel c-MET-based CTC detection platform. Mol. Cancer Res. 2016, 14, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Stanisławska, K.; Stadnik, H.; Nawrocki, M.; Ramlau-Piątek, K.; Juszkat, R.; Drews, M. Long-term survival in a patient with unresectable liver metastases from uveal melanoma treated with transarterial chemoembolization with irinotecan eluting beads—Case report and review of literature. Współczesna Onkol. 2017, 3, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Valeriote, F.A. The use of cell kinetics in the development of drug combinations. Pharmacol. Ther. 1979, 4, 1–33. [Google Scholar] [CrossRef]
- Malvicini, M.; Aquino, J.; Mazzolini, G. Combined Therapy for gastrointestinal carcinomas: Exploiting synergies between gene therapy and classical chemo-radiotherapy. Curr. Gene Ther. 2015, 15, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Inoue, T.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. Cell diameter measurements obtained with a handheld cell counter could be used as a surrogate marker of G2/M arrest and apoptosis in colon cancer cell lines exposed to SN-38. Biochem. Biophys. Res. Commun. 2013, 434, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Abu-Sanad, A.; Wang, Y.; Hasheminasab, F.; Panasci, J.; Noë, A.; Rosca, L.; Davidson, D.; Amrein, L.; Sharif-Askari, B.; Aloyz, R.; Panasci, L. Simultaneous inhibition of ATR and PARP sensitizes colon cancer cell lines to irinotecan. Front. Pharmacol. 2015, 6, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhao, X.; Lee, L.J.; Lee, R.J. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009, 11, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; He, X.; Li, F.; Pan, H.; Huang, X.; Wen, X.; Zhang, H.; Li, B.; Ge, S.; Xu, X.; et al. The miR-181 family promotes cell cycle by targeting CTDSPL, a phosphatase-like tumor suppressor in uveal melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 15–32. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Oligonucleotides | Sequence |
|---|---|---|
| 1 | DNA-34a | 5′-TGGCAGTGTCTTAGCTGGTTGTTAAAAA-3′ |
| 2 | DNA-182 | 5′-TTTGGCAATGGTAGAACTCACACTAAAAA-3′ |
| 3 | DNA-137 | 5′-TTATTGCTTAAGAATACGCGTAGAAAAA-3′ |
| 4 | DNA-144 | 5′-GGATATCATCATATACTGTAAGAAAAA-3′ |
| 5 | DNA-34a | 5′-TGGCAGTGTCTTAGCTGGTTGTTAAAAA–Thiol-3′ |
| 6 | DNA-182 | 5′-TTTGGCAATGGTAGAACTCACACTAAAAA–Thiol-3′ |
| 7 | DNA-137 | 5′-TTATTGCTTAAGAATACGCGTAGAAAAA–Thiol-3′ |
| 8 | DNA-144 | 5′-GGATATCATCATATACTGTAAGAAAAA–Thiol-3′ |
| 9 | RNA-34a pass | 5′-Thiol–AAAAAArCrArArCrCrArGrCrUrArArGrArCrArCrUrGrCrCrU-3′ |
| 10 | RNA-34a guide | 5′-rUrGrGrCrArGrUrGrUrCrUrUrArGrCrUrGrGrUrUrGrU-3′ |
| 11 | RNA-182 pass | 5′-Thiol–AAAAAArGrUrGrUrGrArGrUrUrCrUrArCrCrArUrUrGrCrCrArArA-3′ |
| 12 | RNA-182 guide | 5′-rUrUrUrGrGrCrArArUrGrGrUrArGrArArCrUrCrArCrArCrU-3′ |
| 13 | RNA-137 pass | 5′-Thiol–AAAAACUACGCGUAUUCUUAAGCAAUAA-3′ |
| 14 | RNA-137 guide | 5′-rUrUrArUrUrGrCrUrUrArArGrArArUrArCrGrCrGrUrArG-3′ |
| 15 | RNA-144 pass | 5′-Thiol–AAAAArCrUrUrArCrArGrUrArUrArUrGrArUrGrArUrArUrCrC-3′ |
| 16 | RNA-144 guide | 5′-rGrGrArUrArUrCrArUrCrArUrArUrArCrUrGrUrArArG-3′ |
| 17 | AS1411 [47] | 5′-GGTGGTGGTGGTTGTGGTGGTGGTGGTTTTTT–Dithiolane-3′ |
| Nanoparticle | Reagents Used in the Preparation |
|---|---|
| AuNP DNA mix | 500 μL DNA mix 2 |
| AuNP siRNA mix | 500 μL siRNA mix |
| AuNP SN38 | 4.06 μL AS1411 (492 μM) + 5 μL SN38 (2 mM in DMSO) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milán Rois, P.; Latorre, A.; Rodriguez Diaz, C.; Del Moral, Á.; Somoza, Á. Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma. Biomimetics 2018, 3, 28. https://doi.org/10.3390/biomimetics3040028
Milán Rois P, Latorre A, Rodriguez Diaz C, Del Moral Á, Somoza Á. Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma. Biomimetics. 2018; 3(4):28. https://doi.org/10.3390/biomimetics3040028
Chicago/Turabian StyleMilán Rois, Paula, Alfonso Latorre, Ciro Rodriguez Diaz, Álvaro Del Moral, and Álvaro Somoza. 2018. "Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma" Biomimetics 3, no. 4: 28. https://doi.org/10.3390/biomimetics3040028
APA StyleMilán Rois, P., Latorre, A., Rodriguez Diaz, C., Del Moral, Á., & Somoza, Á. (2018). Reprogramming Cells for Synergistic Combination Therapy with Nanotherapeutics against Uveal Melanoma. Biomimetics, 3(4), 28. https://doi.org/10.3390/biomimetics3040028