Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants


Abstract
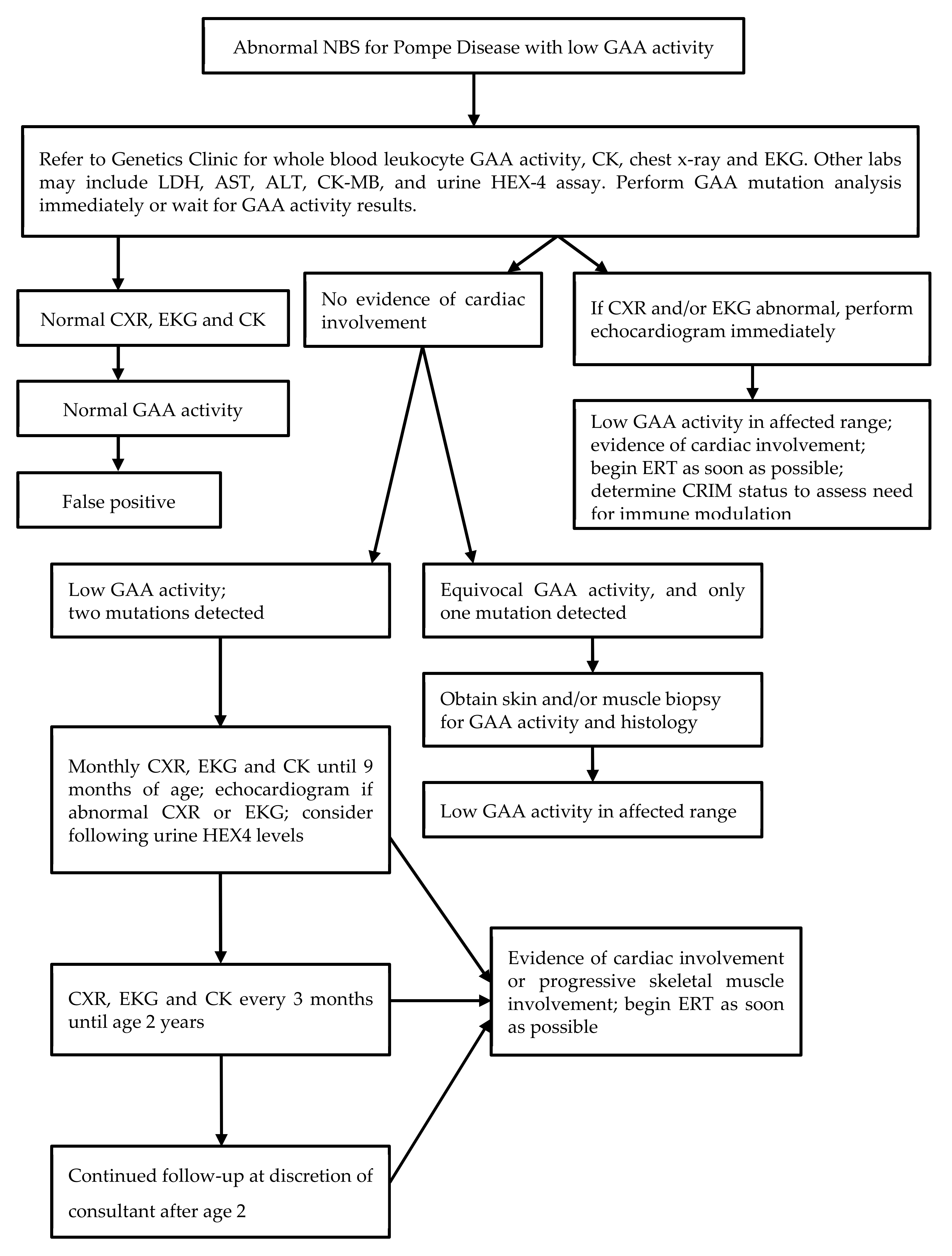
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kishnani, P.S.; Corzo, D.; Leslie, N.D.; Gruskin, D.; van der Ploeg, A.; Clancy, J.P.; Parini, R.; Morin, G.; Beck, M.; Bauer, M.S.; et al. Early treatment with alglucosidase alfa prolongs long-term survival in infants with Pompe disease. Pediatr. Res. 2009, 66, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Goldenberg, P.C.; DeArmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messinger, Y.H.; Mendelsohn, N.J.; Rhead, W.; Dimmock, D.; Hershkovitz, E.; Champion, M.; Jones, S.A.; Olson, R.; White, A.; Wells, C.; et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infants with Pompe disease. Mol. Genet. Metab. 2012, 14, 135–142. [Google Scholar]
- Van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guidelines. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.P.; Piraud, M.; Goldstein, J.L.; Zhang, H.; Rehder, C.; Laforet, P.; Kishnani, P.S.; Millington, D.S.; Bashir, M.R.; Bali, D.S. Assessing disease severity in Pompe disease: The roles of a urine glucose tetrasaccharide biomarker and imaging techniques. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Goldstein, J.L.; Hwu, W.L.; Smith, P.B.; Lee, N.C.; Chiang, S.C.; Tolun, A.A.; Zhang, H.; Vaisnins, A.E.; Millington, D.S.; et al. Baseline urine glucose tetrasaccharide concentrations in patients with infantile- and late-onset Pompe disease identified by newborn screening. JIMD Rep. 2015, 19, 67–73. [Google Scholar] [PubMed] [Green Version]
- Kumamoto, S.; Katafuchi, T.; Nakamura, K.; Endo, F.; Oda, E.; Okuyama, T.; Kroos, M.A.; Reuser, A.J.; Okumiya, T. High frequency of acid alpha-glucosidase pseudodeficiency complicating newborn screening for glycogen storage disease type II in the Japanese population. Mol. Genet. Metab. 2009, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Lee, N.C.; Huang, A.C.; Chen, C.A.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Early detection of Pompe disease by newborn screening is feasible: Results from the Taiwan screening program. Pediatrics 2008, 122, e39–e45. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.C.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Pompe disease in infants: Improving the prognosis by newborn screening and early treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder newborn screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn screening for lysosomal storage disorders in Illinois: The initial 15-month experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.; Cope, H.; Li, J.S.; Kishnani, P.S. Severe cardiac involvement is rare in patients with late-onset Pompe disease and the common c.-32-13T>G variant: Implications for newborn screening. J. Pediatr. 2018, 198, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Kazi, Z.B.; Desai, A.K.; Berrier, K.L.; Troxler, R.B.; Wang, R.Y.; Abdul-Rahman, O.A.; Tanpaiboon, P.; Mendelsohn, N.J.; Herskovitz, E.; Kronn, D.; et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight 2017, 2, e94328. [Google Scholar] [CrossRef] [PubMed]
- Kazi, Z.B.; Desai, A.K.; Troxler, R.B.; Kronn, D.; Packman, S.; Sabbadini, M.; Rizzo, W.B.; Scherer, K.; Abdul-Rahman, O.; Tanpaiboon, P.; et al. An immune tolerance approach using transient low-dose methotrexate in ERT-naïve setting of patients treated with a therapeutic protein: Experience in infantile-onset Pompe disease. Genet. Med. 2019, 21, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Klug, T.; Vermette, L.; Raburn-Miller, J.; Kiesling, J.; Rogers, S. Incidence of 4 lysosomal storage disorders from 4 years of newborn screening. JAMA Pediatr. 2018, 172, 696–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.F.; Yang, C.C.; Liao, H.C.; Huang, L.Y.; Chiang, C.C.; Ho, H.C.; Lai, C.J.; Chu, T.H.; Yang, T.F.; Hsu, T.R.; et al. Very early treatment for infantile-onset Pompe disease contributes to better outcomes. J. Pediatr. 2016, 169, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Reuser, A.J.J.; van der Ploeg, A.T.; Chien, Y.H.; Llerena, J., Jr.; Abbott, M.A.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.J.; et al. GAA variants and phenotypes amount 1,079 patients with Pompe disease; data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Category | Number of Infants Identified |
|---|---|
| Infantile Pompe disease | 3 |
| Late onset Pompe disease | 26 |
| Normal enzyme activity | 234 |
| Carrier 1 | 62 |
| Pseudodeficiency 2 | 39 |
| Phenotype undetermined 3 | 8 |
| Loss to follow-up or refused | 7 |
| Died prior to follow-up 4 | 1 |
| Pending | 15 |
| Case | Genotype | Phenotype | GAA Activity a Result (nl) | CK (IU/L) Result (nl) | Urine Glc4 or Hex4 b Result (nl) | Cardiac Findings | Other Clinical Findings |
|---|---|---|---|---|---|---|---|
| 1 | c.2560C>T, c.1211A>T, c.2161G>C | IOPD | 0.02 (>3.0) | 1064 (35–232) | NR | HCM | Hypotonia |
| 2 | c.1437+1G>A, c.2227C>T | IOPD | 0.8 (>3.88) | 566 (32–250) | NR | HCM | Hypotonia; Motor delay |
| 3 | c.2560C>T, c.2459_2461del | IOPD | 1.6 (>3.88) | 3488 (30–279) | Glc4 14.9 (0.14–1.29) | HCM | Initial hypotonia c |
| 4–16 | c.-32-13T>G homozygous | LOPD | 0.0–2.8 | 153–669 (8/17 elevated) | See footnote d | Normal e | None |
| 17 | c.-32-13T>G, c.1655T>C | LOPD | 0.8 (>3.0) | 550 | Normal | ASD | Mild hypotonia |
| 18 | c.-32-13T>G, c.2238G>C | LOPD | 2.55 (>3.88) | 86 (29–168) | Normal | None | None |
| 19 | c.-32-13T>G, c.1839G>A | LOPD | 1.5 (>3.88) | 641 (30–279) | Glc4 7.59 (0.14–1.29) | None | None |
| 20 | c.-32-13T>G, c.258DPC | LOPD | 0.3 (>3.0) | NR | NR | None | None |
| 21 | c.-32-13T>G, c.2238G>C, c.2065G>A | LOPD | 1.0 (>3.0) | NR | Hex4 41.6 (<20) | RVH on ECG; PFO on echo | None |
| 22 | c.-32-13T>G, c.2297A>G | LOPD f | 2.3 (>3.88) | 168 (55–170) | Glc4 1.21 (0.08–1.37) | Normal | None |
| 23 | c.307T>G, c.1375G>C, c.271G>A | LOPD f | 1.6 (>3.88) | Normal | Glc4 2.0 (1.14–1.29) | Normal | None |
| 24 | c.1637-3_1637-4delinsG, c.1831G>A | LOPD | 2.4 (>3.88) | 93 (30–279) | Glc4 12.98 (0.14–1.29) | Normal | None |
| 25 | c.-32-12T>G, c.2219-2220delTG | LOPD g | 2.0 (>3.88) | 555 (30–279) | Glc4 11.79 (0.14–1.29) | PFO | Hypotonia; gross motor delay |
| 26 | c.2238G>C, c.2242dupG | LOPD f | 2.9 (>3.88) | 142 (30–279) | Glc4 6.54 (0.14–1.29) | Normal | None |
| 27 | c.2173delC, c.858+17-858+23delCGGGGCGG | LOPD | 2.9 (>3.88) | 272 (30–279) | NR | Normal | None |
| 28 | c.1121G>T, c.885C>T | LOPD | 0.3 (>3.0) | NR | NR | Normal | None |
| 29 | c.307T>G, c.525delT | LOPD | 0.5 (>3.0) | 193 (55–170) | NR | Normal | None |
| 30 | c.655G>A, c.1418G>C | UND | 3.0 (>7.4) | 73 (39–308) | Hex4 11.2 (<20) | PFO | None |
| 31 | c.525delT, c.265C>T | UND f | 2.9 (>3.88) | 74 (30–279) | Glc4 6.76 (0.14–1.29) | PFO h | None |
| 32 | c.1942G>A, c.1346C>T, c.2065G>A, c.1726G>A | UND | 0.2 (>3.0) | 167 (39–308) | NR | Normal | None |
| 33 | c.664G>A, c.1346C>T | UND | 0.0 (>3.88) | 101 (30–279) | NR | Normal | None |
| 34 | c.726G>A, c.1357G>A | UND | 0.7 (>3.0) | NR | NR | Normal | None |
| 35 | c.1631T>A, c.2509C>T, c.2065G>A | UND | UND | 0.7 (>3.0) | NR | NR | Normal |
| 36 | c.307T>G, c.265C>T | UND | 2.1 (>3.88) | 152 (30–279) | NR | NR | Normal |
| 37 | c.1781G>A, c.1194+3G>C | UND | 3.5 (>3.88) | 571 (30–279) | Glc4 3.46 (0.14–1.29) | Normal | None |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burton, B.K.; Charrow, J.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hitchins, L.; Hickey, R.; Christensen, K.M.; Groepper, D.; et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int. J. Neonatal Screen. 2020, 6, 4. https://doi.org/10.3390/ijns6010004
Burton BK, Charrow J, Hoganson GE, Fleischer J, Grange DK, Braddock SR, Hitchins L, Hickey R, Christensen KM, Groepper D, et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. International Journal of Neonatal Screening. 2020; 6(1):4. https://doi.org/10.3390/ijns6010004
Chicago/Turabian StyleBurton, Barbara K., Joel Charrow, George E. Hoganson, Julie Fleischer, Dorothy K. Grange, Stephen R. Braddock, Lauren Hitchins, Rachel Hickey, Katherine M. Christensen, Daniel Groepper, and et al. 2020. "Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants" International Journal of Neonatal Screening 6, no. 1: 4. https://doi.org/10.3390/ijns6010004
APA StyleBurton, B. K., Charrow, J., Hoganson, G. E., Fleischer, J., Grange, D. K., Braddock, S. R., Hitchins, L., Hickey, R., Christensen, K. M., Groepper, D., Shryock, H., Smith, P., Shao, R., & Basheeruddin, K. (2020). Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. International Journal of Neonatal Screening, 6(1), 4. https://doi.org/10.3390/ijns6010004