1. Introduction
The world of multi-parameter rare cell analysis is changing. In a move to overcome the restrictions of traditional microscopic analyses using fluorophore-labeled antibodies—which are limited to visualizing three to four different cellular markers at a time—new technologies keep emerging that pledge ever-more analytical depth and improved data accessibility. Despite proving resourceful in research settings, most of them are yet to make a notable impact on the clinical routine.
Even flow cytometry, the only analytic tool with multi-parameter capabilities that has achieved significant clinical cut-through to date, comes with a suite of constraints that need improvement—not least its inability to produce the morphological information available via conventional microscopic analyses. Despite being capable of imaging 20 and more fluorophore-labeled antibodies simultaneously, its relatively high detection limit also restricts the use of flow cytometry in rare cell detection [
1].
Other emerging multi-parameter approaches to rare cell analysis, such as imaging flow cytometry [
2,
3], mass spectrometry-based analysis [
4,
5], and multi-epitope-ligand-cartography (MELC) [
6,
7], have recently shown potential to address some of these shortcomings, but in turn require relatively expensive additional equipment and sophisticated downstream processing of digitally acquired data—disadvantages that are still holding them back from widespread clinical adoption.
As such, next-generation fluorescence microscopy may be considered the most promising method to analyze rare cell populations in everyday clinical settings, especially given the ongoing development of new fluorophores to label antibodies and the generation of new fluorescent proteins.
One way of multiplexing larger numbers of fluorophores in the microscope is spectral imaging followed by linear unmixing. Since this technology relies on the acquisition of spectral image stacks separated by no more than 2–10 nm, it requires tunable excitation light sources and adjustable emission detection windows. It may also result in very large image data sets and lengthy image acquisition and analysis processes.
One slightly more practical variant using the standard lasers of a confocal microscope as the excitation light source has recently been presented by Gerner et al. [
8]. Multi-color laser scanning confocal immunofluorescence microscopy demonstrated five-laser, line-based co-staining of six different fluorophore-labeled antibodies in single-tissue sections when studying murine lymph node dendritic cell subpopulations. However, high spillover—or crosstalk—between emission bands still necessitated significant deconvolution, a computationally intensive processing technique to improve image contrast and resolution. The required compensation algorithms have been limiting the technology’s applicability for daily laboratory use.
In 2014, Eissing et al. went on to combine classical narrow stokes shift fluorescent dyes of the Alexa Fluor series and large stokes shift tandem dyes of the Brilliant Violet series, in turn achieving six-color immunofluorescence microscopy on a confocal microscope with just four lasers and no additional image processing. The tandem’s acceptor dye, however, still exhibited crosstalk into neighboring fluorescence channels [
9].
Finally, in 2015, Kijani et al. demonstrated the use of narrow-bandwidth fluorescence filters and standard fluorochromes for six-color immunofluorescence microscopy on a standard widefield fluorescence microscope [
10]. The authors reported zero crosstalk into neighboring channels, albeit only judging qualitatively from the acquired images, with no empirical quantification performed.
What is more, Kijani et al.’s staining process mostly involved secondary fluorophore-labeled antibodies, which not only required laborious staining procedures, but also resulted in large variations in staining intensity for each channel. This, in turn, resulted in hugely different image acquisition times, ranging from less than 100 ms up to several seconds per channel.
Progressing further towards the practical routine use of true multi-color microscopy, we now provide a protocol that avoids secondary antibodies or tandem dyes and includes a standardized method to empirically quantify crosstalk between channels. This optimized set-up results in standardized image acquisition times of 50 ms per channel on an automated widefield fluorescence microscope equipped with a standard high-pressure mercury vapor arc-discharge light source. We demonstrate that carefully selected combinations of up to seven different, primary fluorophore-labeled antibodies and microscope fluorescence filter sets enable the fast and direct visualization of several cell subsets, with minimal crosstalk between fluorescence channels and no downstream application of advanced mathematical algorithms.
2. Materials and Methods
2.1. Cell Lines and Human Blood Samples
A549 lung adenocarcinoma cells were purchased from Cell Line Services (Eppelheim, Germany) and cultured at 37 °C and 5% CO2 in DMEM/HAM’S F-12 (1:1), 2 mM l-glutamine supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (final concentrations). The cells were harvested with 0.25% trypsin. Blood used to spike experiments was obtained from healthy, voluntary donors in our laboratory.
2.2. Erythrocyte Lysis for Isolation of Human Leukocytes
Twenty milliliters of hMX lysis buffer (X-ZELL, Sunnyvale, CA, USA) was added to 5 mL whole blood. The samples were then incubated for 5–7 min (till clear) and washed with 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, 5 mM EDTA and 1% fetal bovine serum at pH 7.4 (PBS/EDTA/FBS) at 400 g for 10 min at room temperature, at the lowest centrifuge acceleration and deceleration. After centrifugation, the leukocytes were re-suspended in 50 µL PBS/EDTA/FBS and counted in a hemocytometer.
2.3. Staining of A549 Cells Spiked into Human Leukocytes
The cultured A549 cells were spiked into leukocytes at a ratio of 1:20 before resuspending the mixtures in 700 µL cytocentrifugation buffer (X-ZELL), and spinning them onto gelatin-coated slides (X-ZELL) in one-well concentrators of a Statspin Cytofuge 2 (Beckman Coulter, Brea, CA, USA) for 10 min at 600 rpm at room temperature. The slides were fixed in cryofixation buffer I (X-ZELL) for 15 min at −25 °C and re-hydrated in cryofixation buffer II (X-ZELL) for 20 min at −2.5 °C.
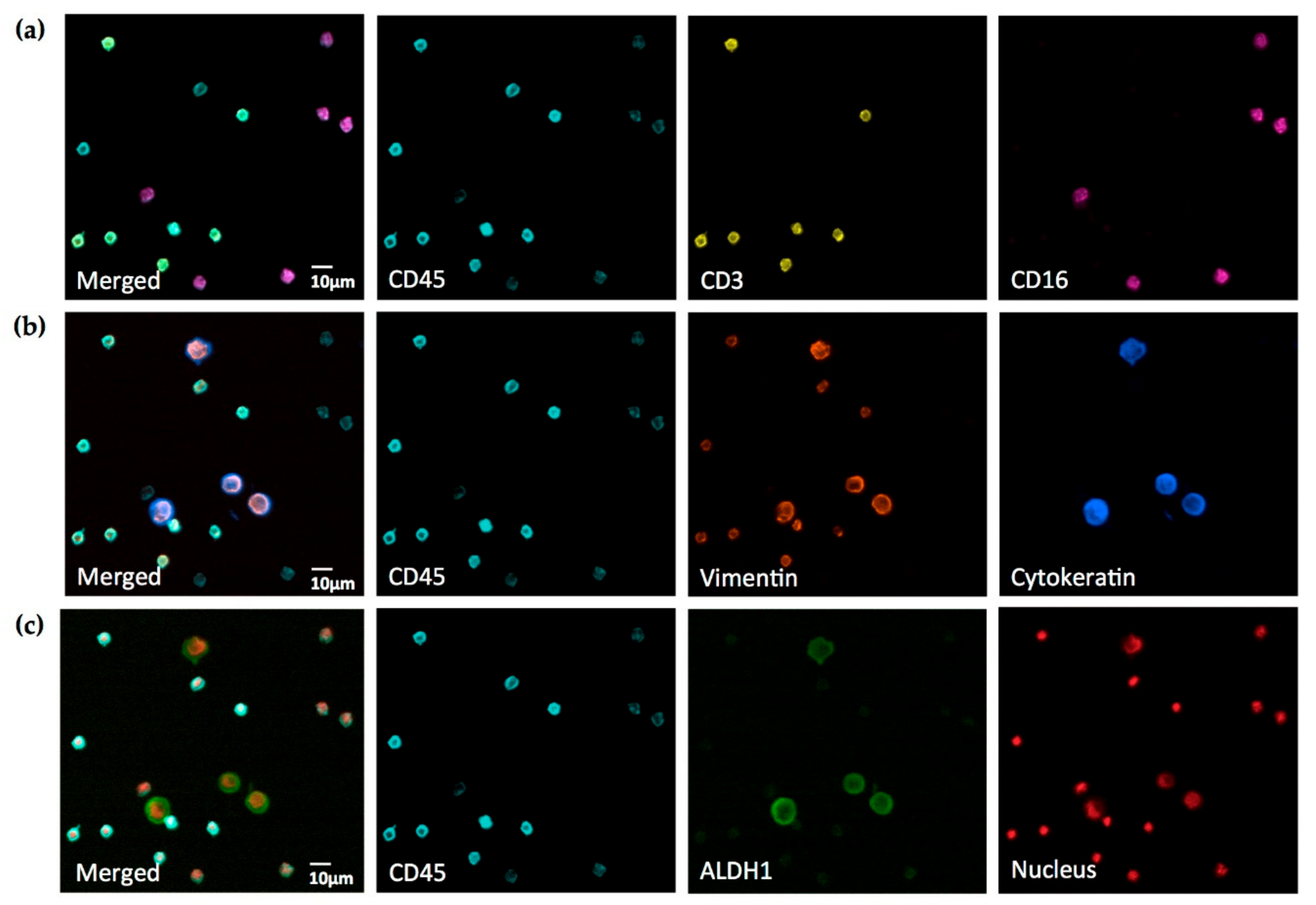
Next, they were mounted on CapGap assemblies (X-ZELL) and inserted in a Cryostainer (X-ZELL). Four microliters of Fc-receptor blocking reagent (Biolegend, San Diego, CA, USA) in 95 µL blocking buffer (X-ZELL) was applied, followed by cocktails of fluorophore-conjugated antibodies in antibody binding buffer as indicated. Antibodies and nuclear dye were as follows: anti-cytokeratin Brilliant Violet 421 (clone: CAM5.2, BD Bioscience, San Jose, CA, USA); anti-CD45 Brilliant Violet 480 (clone: HI30, BD Bioscience); anti-ALDH1 Alexa Fluor 488 (clone: EP1933Y, Abcam, Cambridge, UK); anti-CD3 R-Phycoerythrin (PE) (clone UCHT1, exbio, Prague, Czech Republic); anti-vimentin Alexa Fluor 488, PE and Alexa Fluor 594 (all: clone: EPR3776, Abcam); anti-CD16 PerCp (peridinin- chlorophyll protein complex; clone: 3G8, exbio); and DRAQ5 DNA dye (eBioscience, San Diego, CA, USA). Incubation times were 45 and 60 min for blocking and staining, respectively. After staining, the slides were released from the CapGap clips and coverslipped with 25 µL mounting buffer (MB I) containing DRAQ5 DNA dye.
2.4. Widefield Fluorescence Microscopy
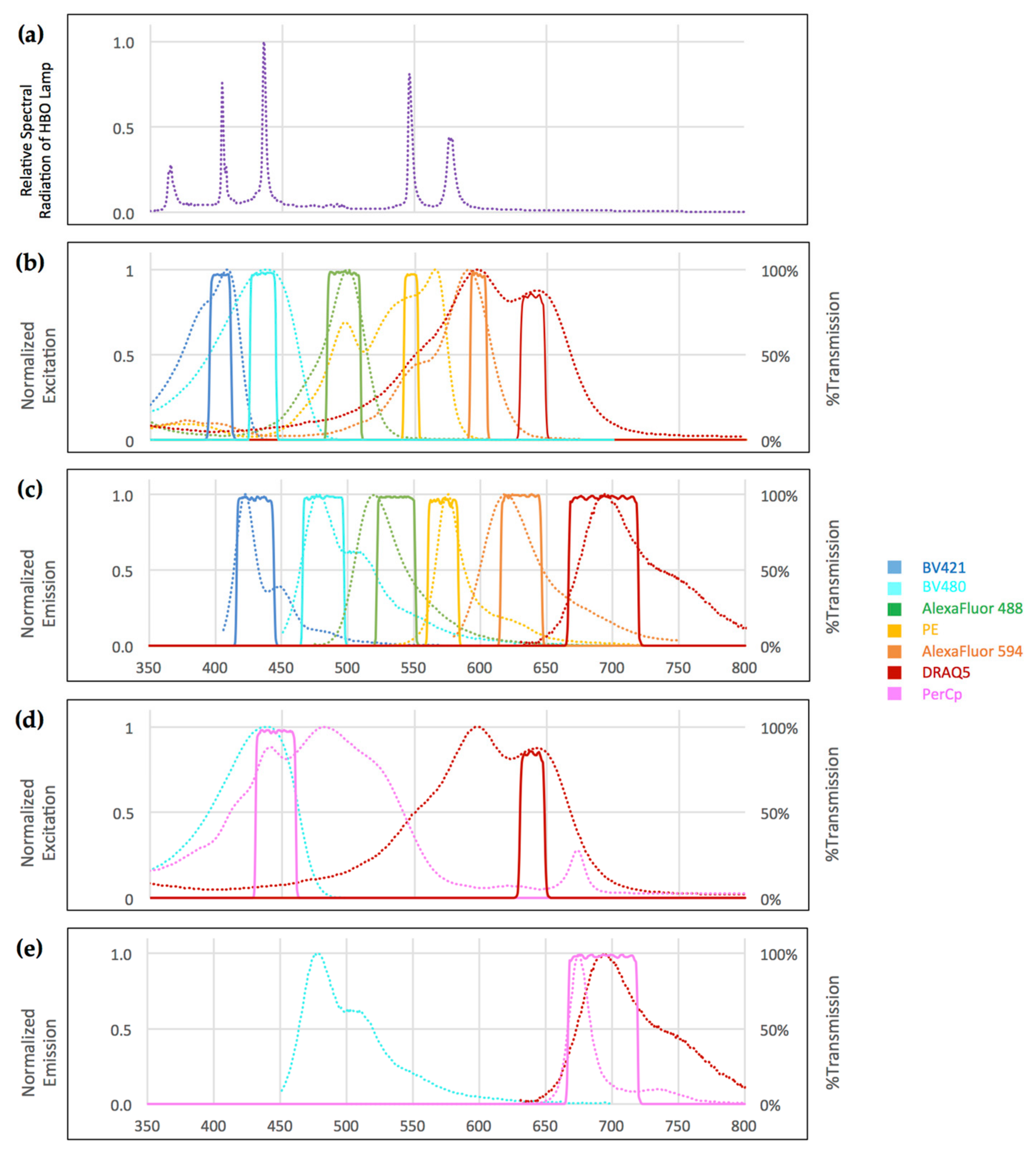
All slides were analyzed on an automated DM6000B widefield fluorescence microscope equipped with an eight-position filter turret, a 100 W high-pressure mercury vapor arc-discharge light source (Leica, Wetzlar, Germany) and an Orca R2 CCD camera (Hamamatsu, Tokyo, Japan). Images were recorded and analyzed using a 40×/0.75 Fluotar objective (Leica) and MicroManager 1.4 software (MicroManager, San Francisco, CA, USA). The filter sets used for each fluorophore and the optical set-up of the microscope are shown in
Table 1 and
Figure 1, respectively. The exposure time was set to 50 ms per fluorescence channel at a 1344 × 1024 pixel resolution. For the experimental set-up, we used spectral data in ASCII (American Standard Code for Information Interchange) format and the fluorescence spectra viewer, both from Chroma Technology Corporation (Bellows Falls, VT, USA) [
11].
2.5. Measuring Integrated Fluorescence Intensity and Determining Crosstalk
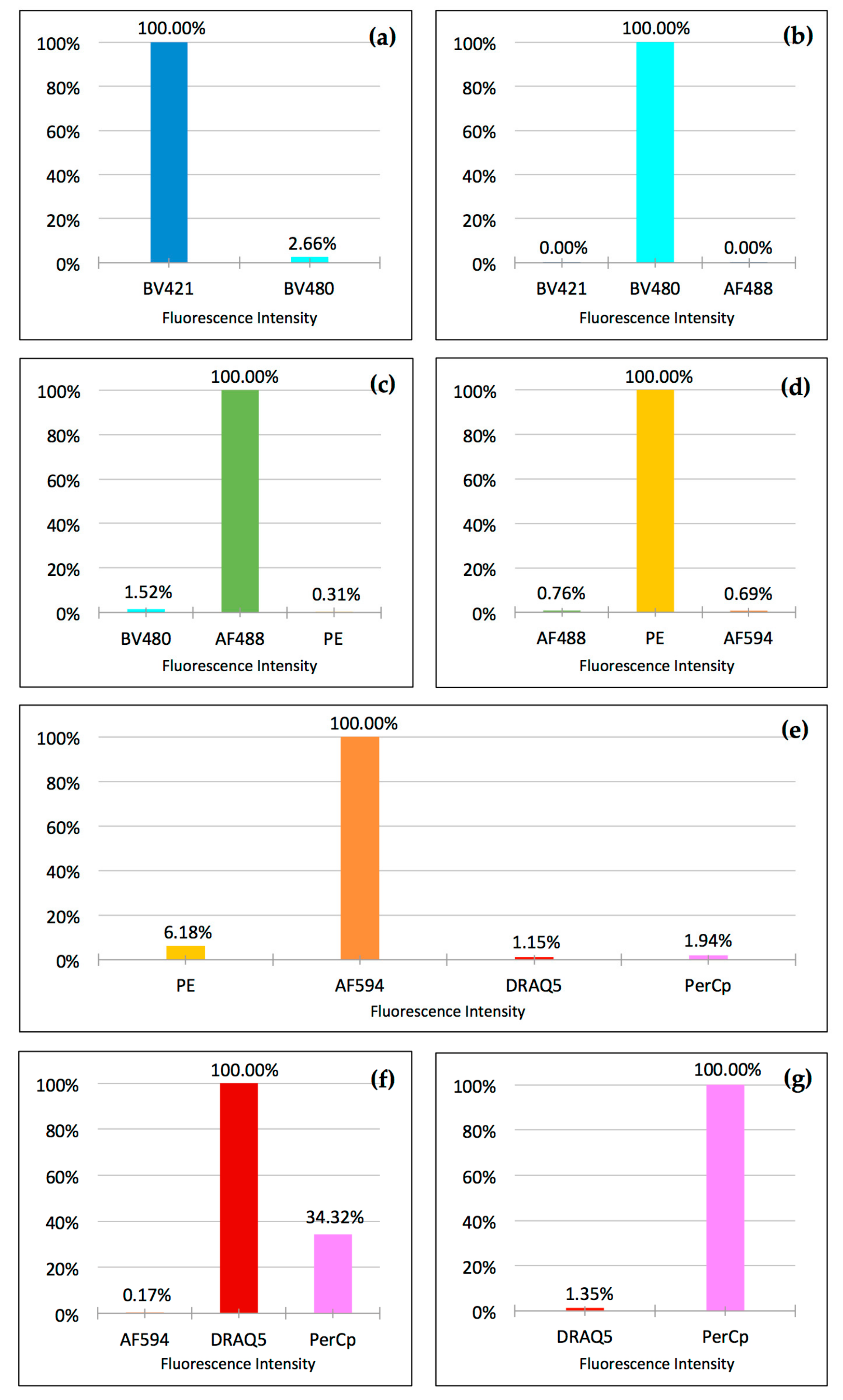
To quantify potential crosstalk, we measured the integrated fluorescence density in ImageJ (as described in the software documentation) [
12]. After selecting regions of interest (ROI) containing fluorescent events (cells), we used the software’s ROI manager to measure fluorescence density and subtracted the background. The signal intensity of the main channel was normalized to “1”. Signal intensity in the adjacent channels then reflected crosstalk and was expressed as a percentage of the intensity of the main channel.
4. Discussion
By imaging human cancer cells spiked into human leukocytes, we were able to demonstrate that up to seven fluorophores can be employed on a standard widefield fluorescence microscope and detected specifically with negligible crosstalk. The resulting seven-color images can be acquired in less than 10 s per seven-channel picture, providing new insights into the co-localization of various cell antigens in human blood. Importantly, the fluorophores employed in this study are standard reagents in flow cytometry and available conjugated to a very wide range of antibodies, which will facilitate the design of suitable antibody panels for most research questions.
The simple staining protocol—which takes advantage of primary fluorophore-conjugated flow-cytometry antibodies—facilitates titration to obtain similar emission brightness in all channels, enabling very fast image acquisition times. The set-up for seven colors also offers additional advantages over the previously published set-up for six-color imaging [
10]. In addition, we offer a reproducible way to empirically quantify crosstalk between neighboring channels, which should assist researchers to objectively optimize their own multi-color microscopy set-up.
Compared to other potent cell analysis tools, our approach also enables the observation of several cell subsets and their morphologies—in turn revealing important information about the presence of an antigen at the single cell level, as well as its extracellular, intracellular, or nuclear protein distribution and structure. Characterizing cells in such detail on a single slide can help avoid common interpretation errors, which may occur when analyzing data retrieved via flow cytometry or other methods that do not disclose information regarding cell morphology.
Our experiments also demonstrated that a carefully selected combination of filter sets and fluorophores enables the crosstalk-free visualization of cells fixed and stained on standard slides. The only significant exception was—as expected from our set-up experiments—DRAQ5 talking into the PerCp channel and, to a much lower degree, AlexaFluor 594 talking into the PE channel. However, in actual seven-color staining, the signal-to-noise ratios were sufficiently large to obtain clear images for analysis by only adjusting the image brightness (
Figure 4). Interestingly, when using the eyepiece of the microscope, dim crosstalk could be recognized in the PE channel, but not in the PerCp channel. This seemingly contradictory finding can be explained by the fact that the human eye has a much superior dynamic contrast ratio over any modern camera in the visible light spectrum (PE channel), but loses this advantage when approaching the near-infrared spectrum (PerCp channel) [
14]. Also, while low-expression antigens may pose challenges by reducing the signal-to-noise ratio, the distinct morphology of nuclei and cellular structures will still enable researchers to distinguish between crosstalk and a specific signal in the case of nuclear staining talking into a membrane antigen channel.
The key to selecting the correct filter set combinations lies in the fluorophores’ distinct slopes of excitation and emission spectra. We were also able to avoid tandem dyes, which tend to exhibit fluorescence bleed-through into the emission channel of the donor-dye and unwanted excitation in the channel of the acceptor dye, as observed in our laboratory and by other authors [
9].
It is important to note that the excitation maxima of most fluorophores fall onto, or close to, the emission peaks of classical high-pressure mercury bulbs. In our set-up, the mercury 405 nm peak coincides with the Brilliant Violet 421 excitation maximum, while the 436 nm peak matches with the Brilliant Violet 480 maximum and the PerCp maximum. The 546 nm peak coincides with the PE maximum, and the 579 nm peak is close to the Alexa Fluor 594 maximum. Taking advantage of this observation facilitates the use of very narrow-bandwidth excitation filters.
In our set-up, the only exceptions were the excitation spectra peaks of Alexa Fluor 488 and DRAQ5, which do not coincide with mercury emission peaks. Therefore, it is advisable to use Alexa Fluor 488 for highly expressed antigens. DRAQ5, on the other hand, binds to DNA stochiometrically, and we found that the concentration can conveniently be up-titrated to give robust visibility in our system. In fact, far-infrared dyes such as AlexaFluor 750 or AlexaFluor 800 could be added to the panel to add an eighth channel. In standard laboratory settings, however, the relatively low spectral radiation of the mercury bulb in this range of the spectrum and limited availability of antibody conjugates somewhat reduce the applicability of these fluorophores.
Taking these physical and physiochemical limitations into account, excitation filters can therefore be chosen with very narrow bandwidths around or close to mercury emission and fluorophore excitation peaks. This minimizes cross-excitation of adjacent fluorophores and—in combination with corresponding narrow-bandwidth emission filters—effectively avoids crosstalk (
Figure 2 and
Figure 3 and
Table 1).
Accordingly, we conclude that if the correct filters and fluorophores are used in combination, there is no need to apply advanced compensation correction algorithms (as recently suggested for another method in histocytometry [
8]), which are undesirable as they tend to reduce the image’s signal-to-noise ratio when subtracting the grey values of overlapping emission spectra. In microscopic imaging, a good signal-to-noise ratio is essential to reproducibly detect faint signals of low-expressed antigens and ensure a high sensitivity.
With that in mind, the lack of excitation intensity of the mercury bulb in the 480–510 nm region and the far infrared region somewhat limits the detection of low-expressed antigens at the moment. However, increasing the lamp wattage or switching to high-powered metal-halide or Xenon light sources offering emission spectra that are more consistent could remedy this shortcoming when required.
Compared to flow cytometry—widely considered the most application-rich single-cell characterization technology to emerge over the past 40 years—our approach does not forego crucial information on cell morphology and keeps samples intact for repeat analysis. Cells used in flow cytometry, meanwhile, are considered lost after being analyzed once.
The same is true for the more advanced image flow cytometry approach, which also requires more sophisticated equipment than our widefield microscope-based solution, and with it, on-going maintenance and management by a highly trained specialist.
Other analytic tools, such as confocal immunofluorescence microscopy and MELC [
6,
7,
9]—where multiple fluorescein isothiocyanate (FITC)- or PE-labeled antibodies are used in serial application on tissues or single cells—are also restricted by the need for expensive equipment, and may also require sophisticated downstream processing of digitally acquired data. In a move to maximize clinical utility, our approach successfully avoids all of these constraints.







{kind=link}
{kind=link}
{kind=link}
{kind=link}