
A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
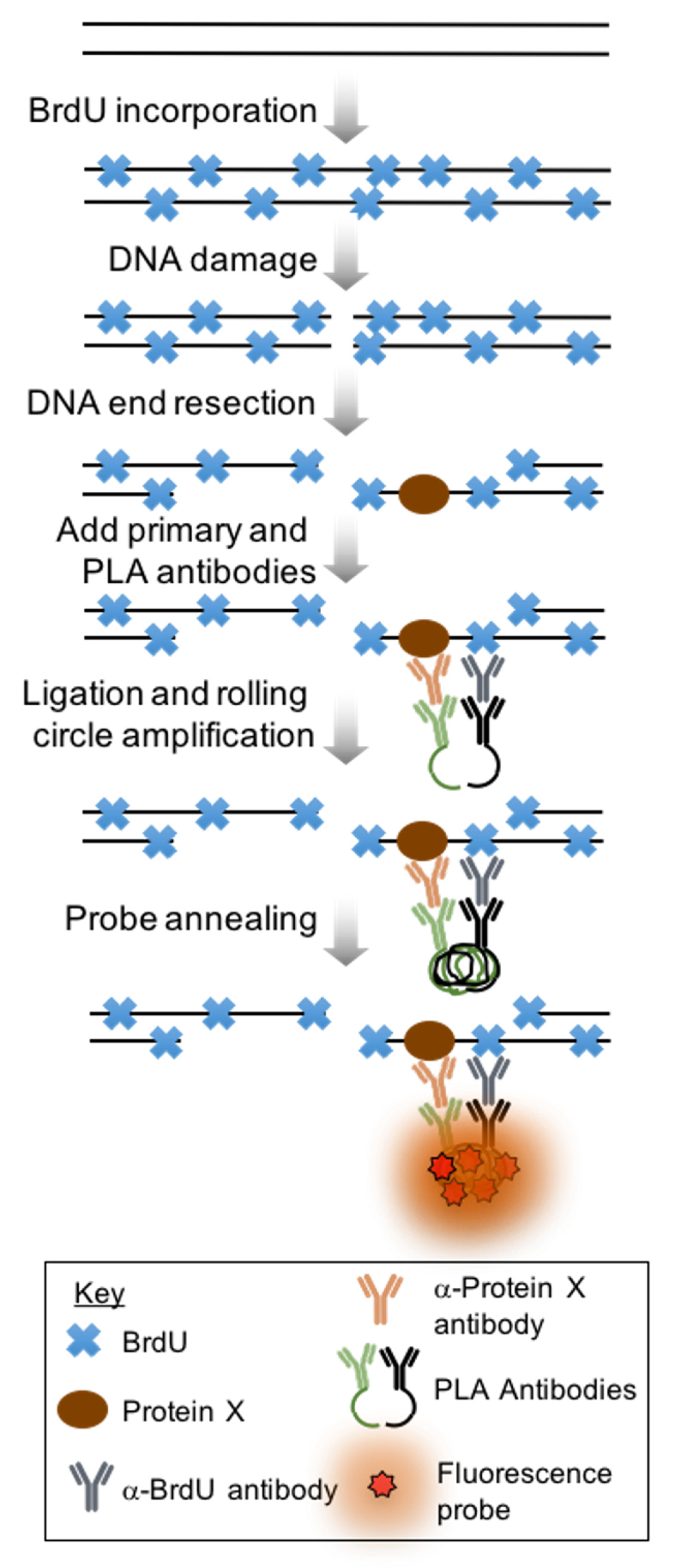
:1. Introduction
2. Experimental Design
2.1. Materials
- 6 cm tissue culture dish (TPP, MIDSCI, Valley Park, MO, USA, Cat. No.: TP93060);
- 24 well tissue culture plates (TPP, MIDSCI, Valley Park, MO, USA, Cat. No.:TP92424;
- 12 mm round glass cover slips (Electron Microscopy Sciences, Hatfield, PA, USA, Cat. No.: 72196-12);
- 0.01% Poly-L-Lysine (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: A-005-C);
- U-2 OS cells (ATCC, Manassas, VA, USA, Cat. No.: HTB-96);
- BrdU (BD Biosciences, Franklin Lakes, NJ, USA, Cat. No.: 550891);
- Bleomycin sulfate (Enzo Life Sciences, Farmingdale, NY, USA, Cat. No.: BML-AP302-0010);
- Triton-X;
- Formaldehyde;
- 10X PBS (Lonza, Thermo Fisher Scientific, Waltham, MA, USA, Cat. No.: BMA51226);
- Purified mouse anti-BrdU clone 3D4 (BD Biosciences, Franklin Lakes, NJ, USA, Cat. No.: 555627);
- Rabbit anti-RPA70 (Cell Signaling Technology, Danvers, MA, USA, Cat. No.: 2267).
- Duolink® In Situ PLA® Probe Anti-Rabbit PLUS (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: DUO92002);
- Duolink® In Situ PLA® Probe Anti-Mouse MINUS (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: DUO92004);
- Duolink® In Situ Detection Reagents Red (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: DUO92008);
- Duolink® In Situ Wash Buffers, Fluorescence (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: DUO82049);
- Aluminum foil;
- Nuclease-free water;
- Frosted Microscope Slides (Fisherbrand, Thermo Fisher Scientific, Waltham, MA USA, Cat. No.: 12-550-343);
- Prolong Gold Antifade Mountant with DAPI (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA, Cat. No.: P36931).
2.2. Equipment
- 37 °C, 5% CO2 incubator;
- Tissue culture hood;
- Rocker;
- Forceps;
- BioTek Lionheart LX Automated Microscope with Gen5 Software (BioTek Instruments, Winooski, VT, USA);
- Microsoft Excel Software;
- GraphPad Prism Software (GraphPad Software, San Diego, CA, USA);
- ImageJ Software (https://imagej.nih.gov/ij/ (accessed on 25 November 2021));
- CellProfiler Software (www.cellprofiler.org (accessed on 25 November 2021));
- X-ray Irradiator (Rad Source Technologies, Buford, GA, USA, Cat. No.: RS 2000).
3. Procedure
3.1. Cell Culture (3 Days)
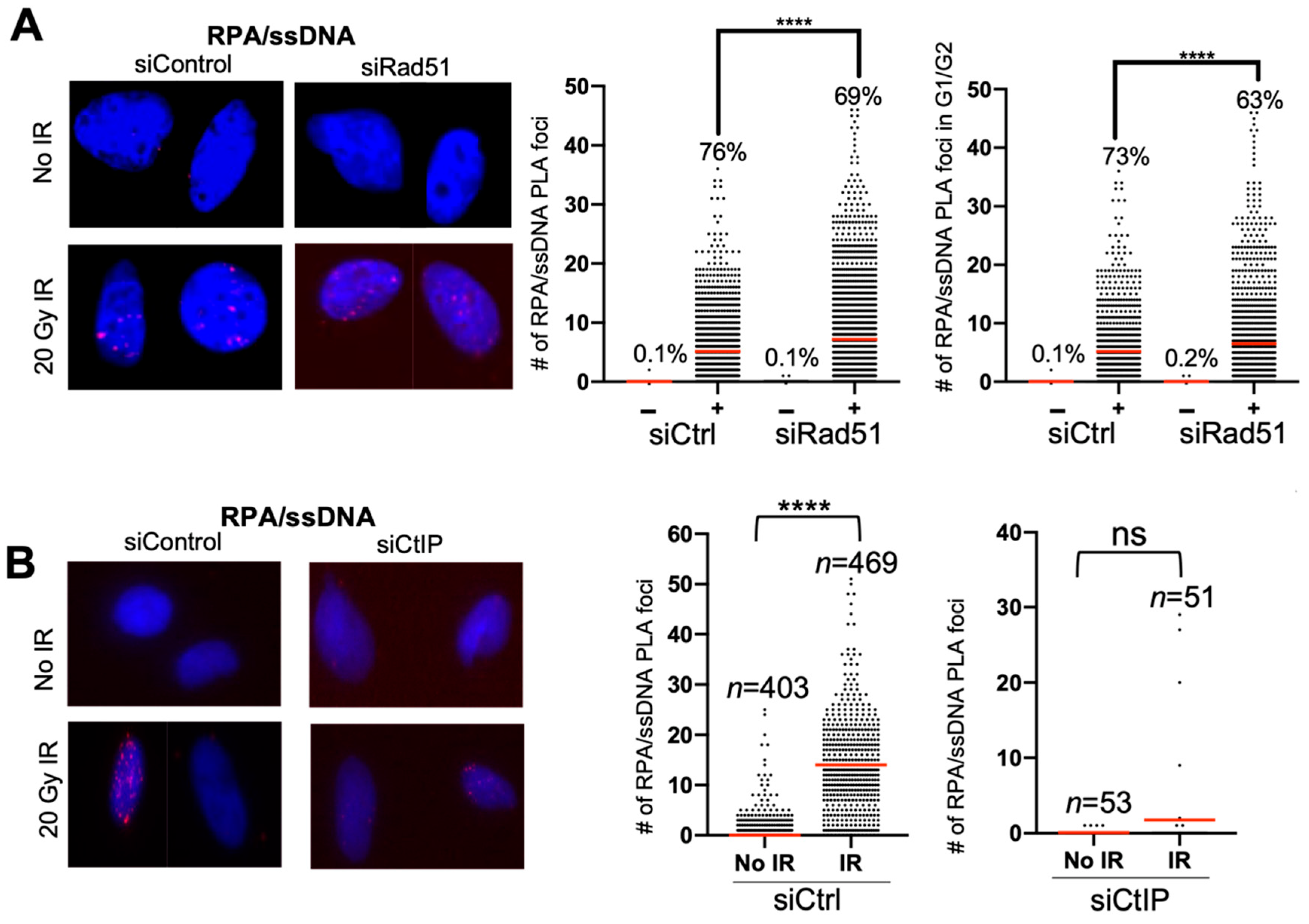
- Optional: perform siRNA knockdowns of a protein of interest if evaluating effect of protein loss on DNA end resection. For example, transfect cells with 5 nM siControl, siRad51, and siCtIP, and incubate for 48–72 h before moving on to step 2.
- Seed 500,000 U-2 OS cells in a 6 cm dish with media containing 10 μM BrdU and place in incubator;
- Approximately 48 h later, place the appropriate number of poly-L-lysine coated glass coverslips into the wells of a 24-well plate;
- Seed 75,000 U-2 OS cells treated with BrdU onto coverslips and incubate for 18–24 h;
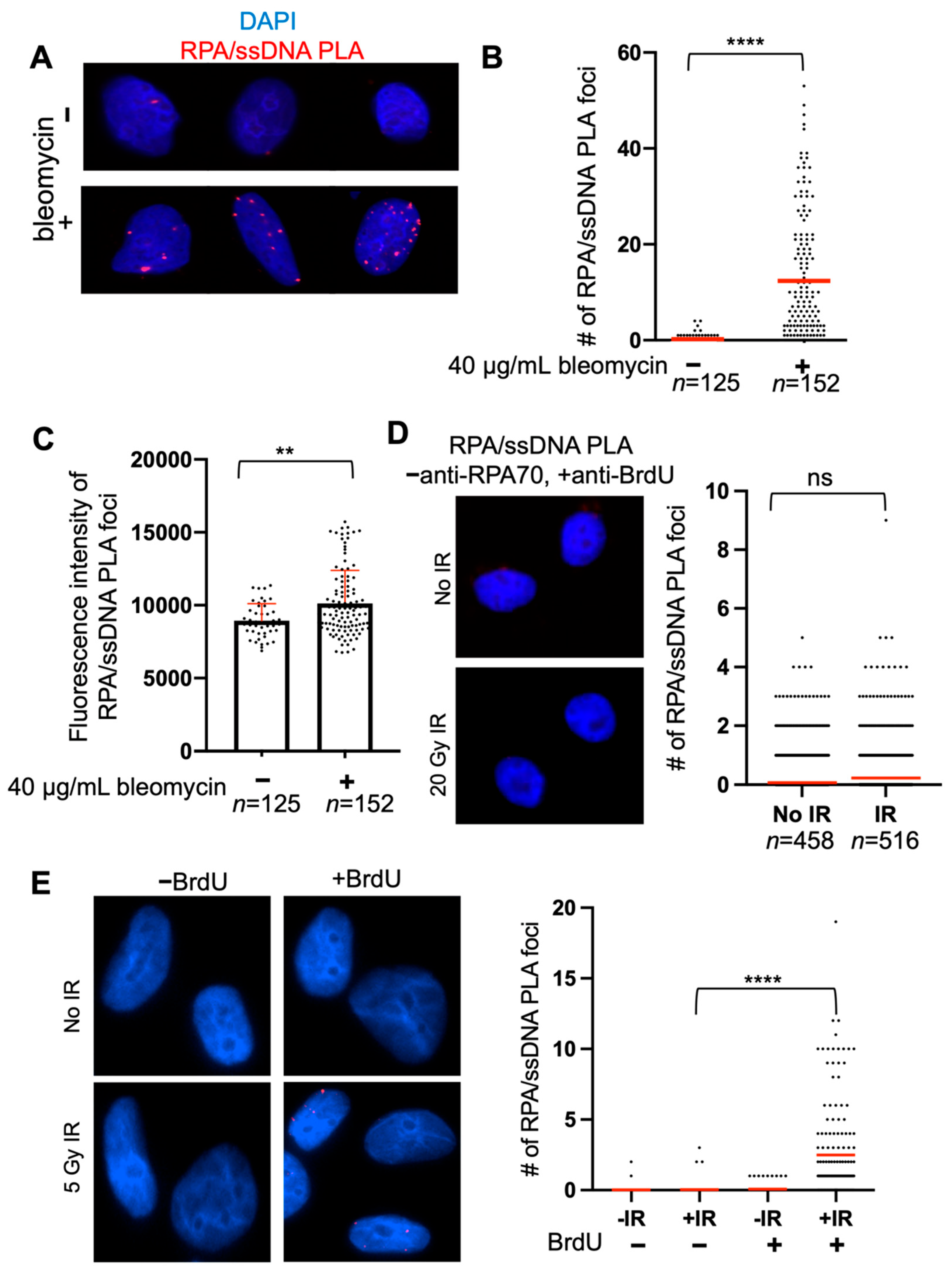
- Induce DNA damage by either treating cells with 40 μg/mL bleomycin or 20 Gray irradiation and allow to recover for 1–8 h.
3.2. Cell Pre-Extraction and Fixation (65 min)
- Wash cells 3 for 5 min in 1X PBS on a rocker at room temperature;
- Pre-extract cells with cold 0.2% Triton-X in PBS for 5 min on a rocker at room temperature;
- Wash cells 3 for 5 min in 1 PBS on a rocker at room temperature;
- Fix cells with 4% formaldehyde in PBS for 15 min on a rocker at room temperature;
- Wash cells 3 for 5 min in 1 PBS on a rocker at room temperature.
 PAUSE STEP Fixed cells can be stored at 4 °C in PBS for up to one week.
PAUSE STEP Fixed cells can be stored at 4 °C in PBS for up to one week.3.3. Primary Antibody Incubation (1 h Plus Overnight)
- Incubate cells with 5 drops of Duolink Blocking Solution at 37 °C for 1 h;
- Dilute primary antibodies in included antibody diluent 1:500;
- Incubate cells in primary antibody overnight at 4 °C on a rocker.
3.4. Proximity Ligation Assay (~4 h 15 min Plus Overnight)
- Wash cells 3 for 5 min in Wash Buffer A on a rocker at room temperature;
- Dilute oligo-conjugated mouse minus and rabbit plus secondary antibodies 1:5 in included antibody diluent;
- Incubate cells at 37 °C for 1 h;
- Wash cells 3 for 5 min in Wash Buffer A on a rocker at room temperature;
- Dilute thawed ligation buffer 1:5 in nuclease-free water, then dilute ligase 1:40;
- Incubate cells with ligation mixture at 37 °C for 30 min;
- Wash cells 3 for 5 min in Wash Buffer A on a rocker at room temperature;
- Dilute thawed amplification buffer 1:5 in nuclease-free water, then dilute polymerase 1:80;
- Incubate cells in amplification mixture for 100 min at 37 °C;
 CRITICAL STEP Protect from light, wrap plates in foil.
CRITICAL STEP Protect from light, wrap plates in foil.- 10.
- Wash cells 2 for 10 min with Wash Buffer B, then 1 for 1 min with 0.01 Wash Buffer B;
- 11.
- Add 7 μL of Prolong Gold Antifade with DAPI onto a glass slide;
- 12.
- Carefully pick up cover slip from the well using forceps and place cell-side down onto glass slide;
- 13.
- Allow cover slips to cure at least overnight in the dark.
3.5. Imaging and Analysis
- Take pictures of slides using DAPI and mCherry channels. At least 100 cells/5 different fields should be imaged. If using the BioTek Lionheart with Gen5 software, pictures of a large area of the slide can be taken and stitched together to create one picture (montage);
- If desired, quantify DAPI fluorescence using Gen5 software, ImageJ, or CellProfiler [18].
- Perform image preprocessing to subtract background fluorescence;
- Adjust primary mask (DAPI) fluorescence threshold and cell size to create appropriate primary mask that accurately circles each nucleus;
- Measure total (integral) fluorescence of each nuclei.
- G0/G1 boundary = Average DAPI Fluorescence − (0.5 (Standard Deviation))
- G2/M boundary = Average DAPI Fluorescence + (0.5 (Standard Deviation))
- S = Values within −/+ 0.5 Standard Deviation
- 3.
- Count PLA foci manually or with a program such as Gen5, CellProfiler, or ImageJ.
- 4.
- If desired, mask PLA foci and measure fluorescence intensity with Gen5, CellProfiler, or ImageJ;
- 5.
- Enter data to be analyzed into an analysis software such as Graphpad Prism, such as # of foci in each nucleus counted. Dot plots can be graphed and the mean, median, and significance of these data can be calculated.
4. Expected Results
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Maser, R.S.; Monsen, K.J.; Nelms, B.E.; Petrini, J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997, 17, 6087–6096. [Google Scholar] [CrossRef] [Green Version]
- de Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lisby, M.; Symington, L.S. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol. Cell 2013, 50, 589–600. [Google Scholar] [CrossRef] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Kowalczykowski, S.C. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 2002, 277, 31663–31672. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Forment, J.V.; Walker, R.V.; Jackson, S.P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytom. A 2012, 81, 922–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, R.; Wijnhoven, P.; le Sage, C.; Tjeertes, J.; Galanty, Y.; Forment, J.V.; Clague, M.J.; Urbé, S.; Jackson, S.P. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 2014, 16, 1016–1026. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, B.; Tomimatsu, N.; Burma, S. Immunofluorescence-based methods to monitor DNA end resection. Methods Mol. Biol. 2015, 1292, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; Lindquist, R.A.; Papallo, A.; Sabatini, D.M.; Golland, P.; Carpenter, A.E. CellProfiler Analyst: Data exploration and analysis software for complex image-based screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held, P. Monitoring Cell Cycle Progression in Cancer Cells; Biotek Instruments, Inc.: Winooski, VT, USA, 2018. [Google Scholar]
- Roukos, V.; Pegoraro, G.; Voss, T.C.; Misteli, T. Cell cycle staging of individual cells by fluorescence microscopy. Nat. Protoc. 2015, 10, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Mestre, T.; Carneiro, P.; Sahumbaiev, I.; Seruca, R.; Sanches, J.M. Blue intensity matters for cell cycle profiling in fluorescence DAPI-stained images. Lab. Investig. 2017, 97, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wei, C.; Li, J.; Xing, P.; Zheng, S.; Chen, X. Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. 2017, 49, 655–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.J.; Gibb, B.; Kwon, Y.; Sung, P.; Greene, E.C. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017, 45, 749–761. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Shi, L.Z.; Zhu, Q.; Wu, P.; Zhang, Y.W.; Basilio, A.; Tonnu, N.; Verma, I.M.; Berns, M.W.; Hunter, T. CtIP links DNA double-strand break sensing to resection. Mol. Cell 2009, 36, 954–969. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fowler, F.C.; Tyler, J.K. A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks. Methods Protoc. 2022, 5, 3. https://doi.org/10.3390/mps5010003
Fowler FC, Tyler JK. A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks. Methods and Protocols. 2022; 5(1):3. https://doi.org/10.3390/mps5010003
Chicago/Turabian StyleFowler, Faith C., and Jessica K. Tyler. 2022. "A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks" Methods and Protocols 5, no. 1: 3. https://doi.org/10.3390/mps5010003
APA StyleFowler, F. C., & Tyler, J. K. (2022). A Proximity Ligation Method to Detect Proteins Bound to Single-Stranded DNA after DNA End Resection at DNA Double-Strand Breaks. Methods and Protocols, 5(1), 3. https://doi.org/10.3390/mps5010003