
Interactive Analysis, Exploration, and Visualization of RNA-Seq Data with SeqCVIBE



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset Retrieval and Basic Quality Control
2.2. RNA-Seq Data Preprocessing
3. Implementation
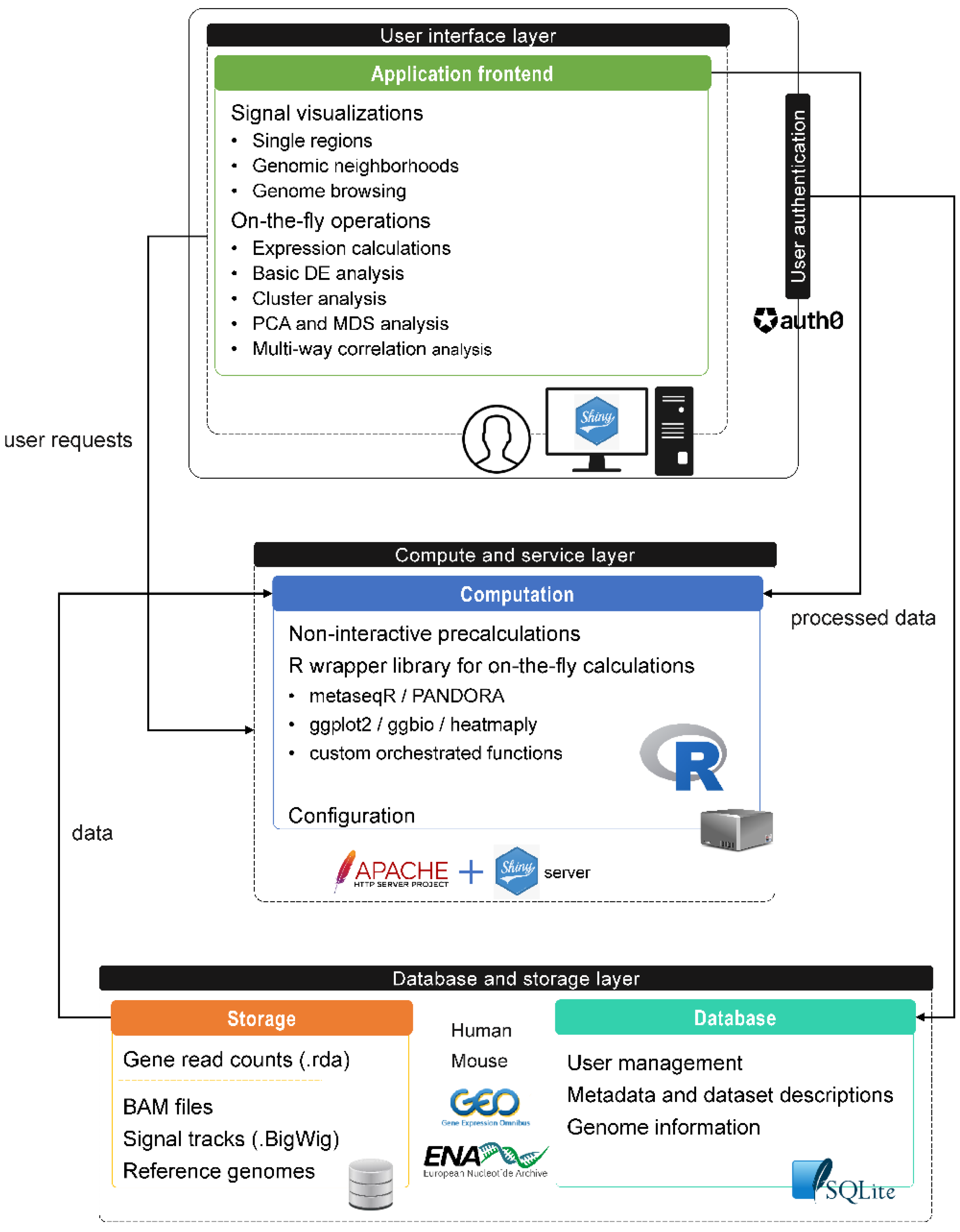
3.1. Overview of SeqCVIBE
- Quality control reports, generated with MultiQC [17].
- Optionally aligned and spliced reads for each sample (BAM files).
- Normalized RNA-Seq signal tracks (BigWig files).
- Precalculated read counts for each exon of each annotated gene/transcript of the reference genome used for read alignment.
- Genomic co-ordinate annotation files for each gene of the reference genome used for read alignment.
3.2. Application Structure
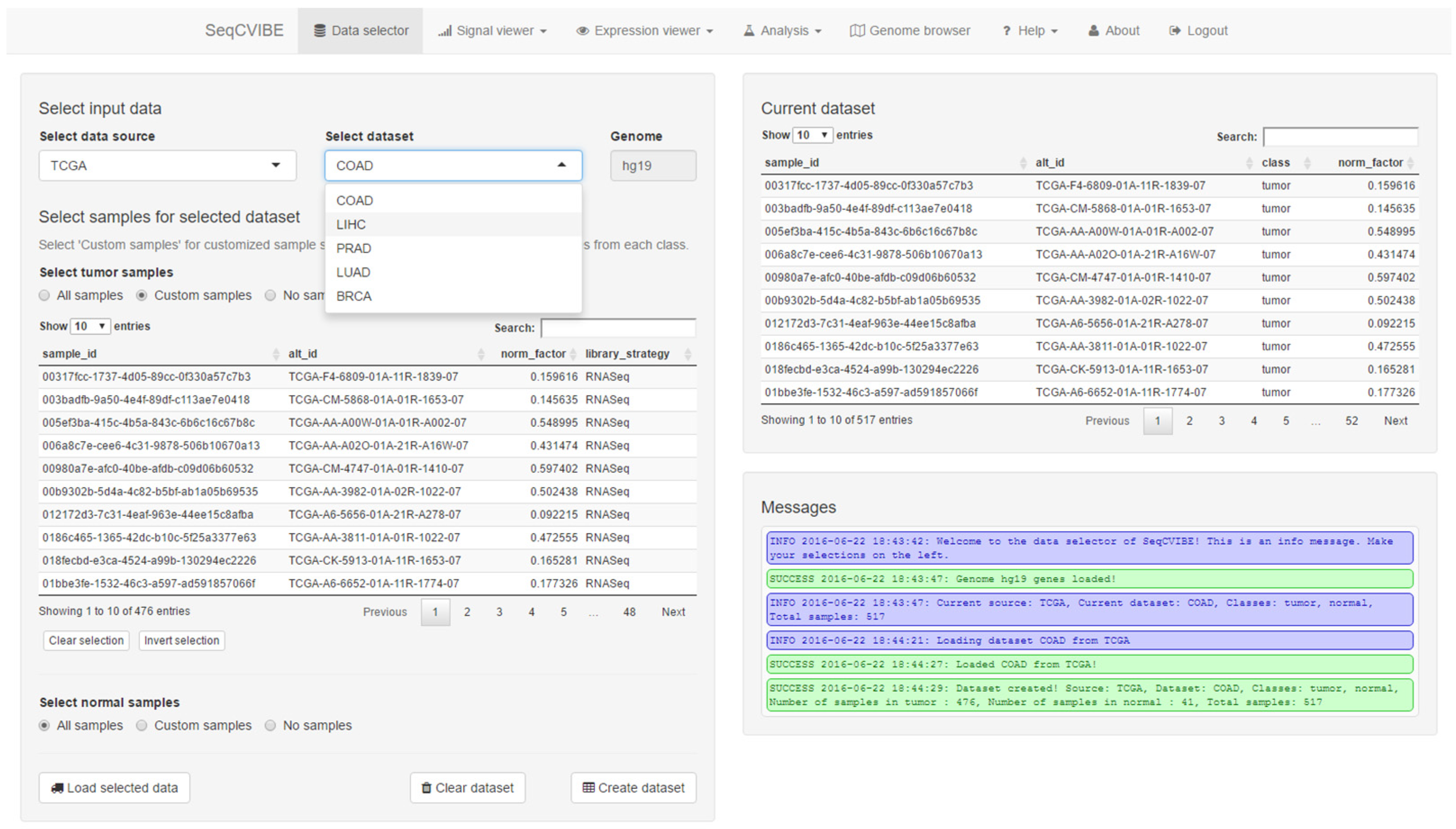
3.2.1. Administrative Functions
- Data sample set management, through the navigation and selection among datasets and samples hosted in SeqCVIBE and the subsequent creation of dataset instances for analysis and querying with the selected biological conditions and samples. Dataset instances can be edited and deleted.
- Analysis instance management, through the ability to work on specific dataset instances, stores the steps as Shiny bookmarks and restores the analytical steps at later times to continue work. Analysis instances can be edited and deleted.
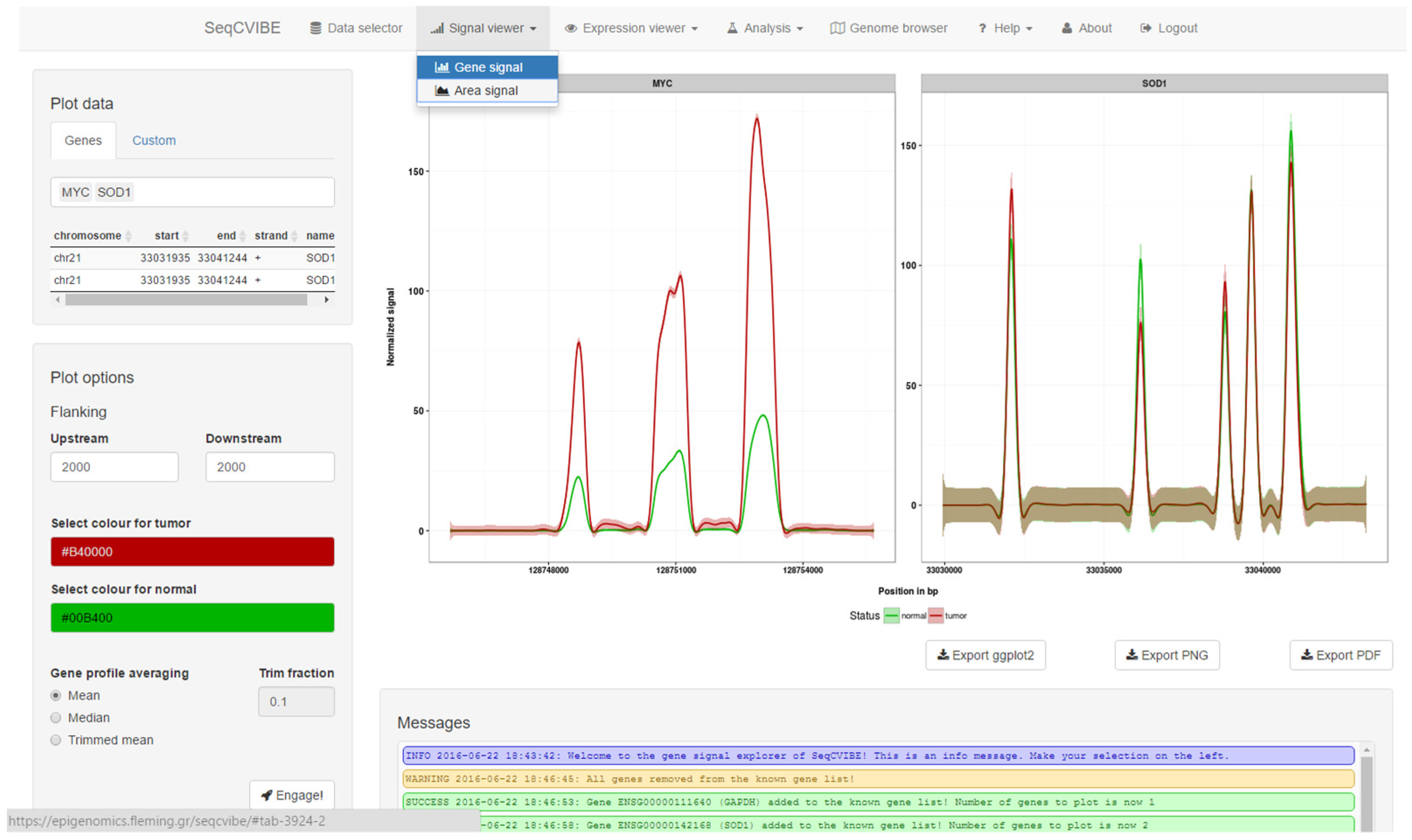
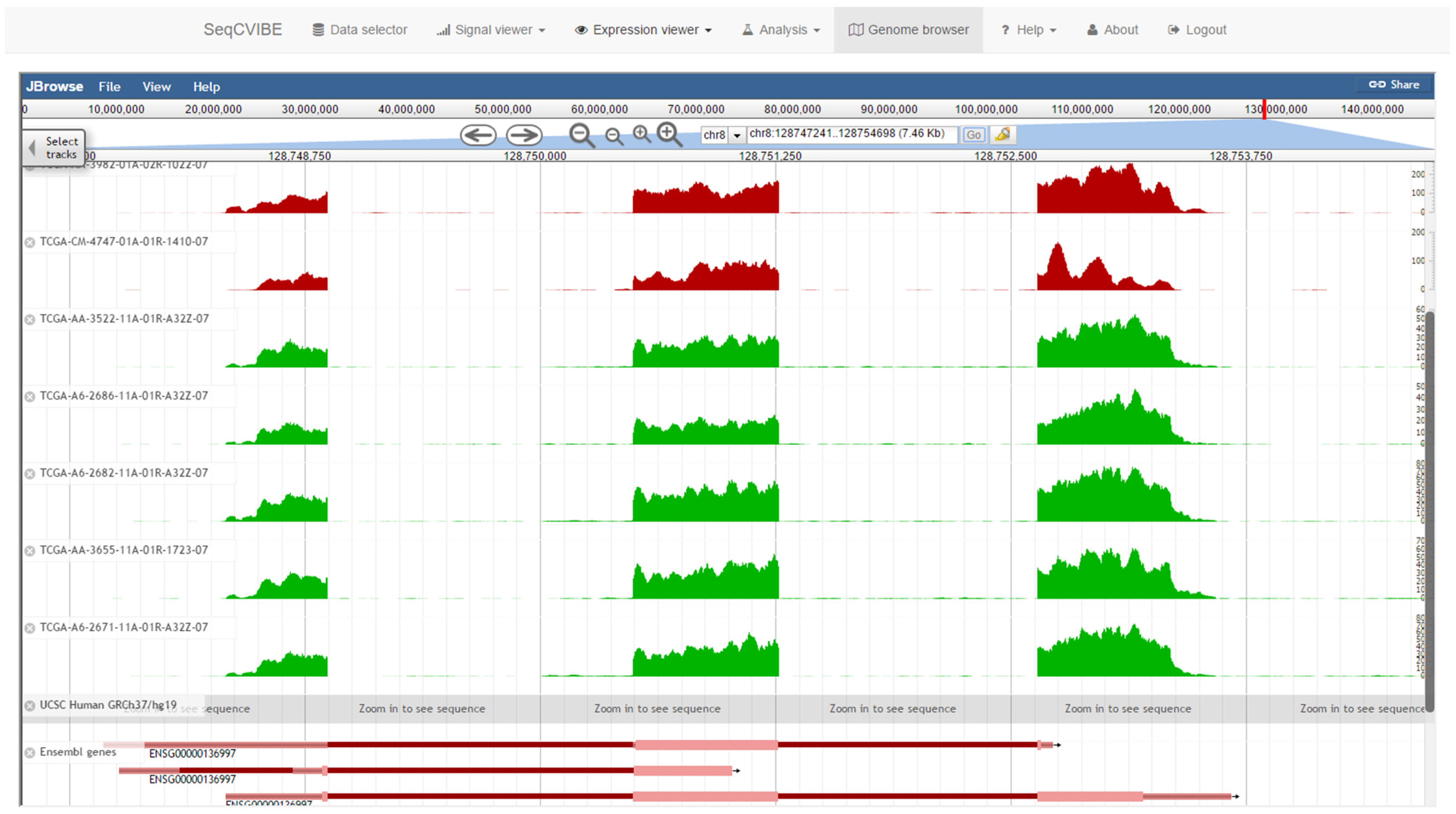
3.2.2. Dynamic RNA-Seq Signal Plots
- Genomic regions within and around known annotated transcripts.
- Genomic regions within and around non-annotated regions where transcription events have been detected via RNA-Seq, such as potentially novel long non-coding RNAs.
- Larger genomic regions within the same chromosome, with the goal of exploring average signals from multiple areas at the same time. This feature resembles the functionalities of a genome browser, but the tracks are averaged, providing a better overview, especially in the case of datasets with many samples. This is achieved with functionalities from the ggbio Bioconductor package [18].
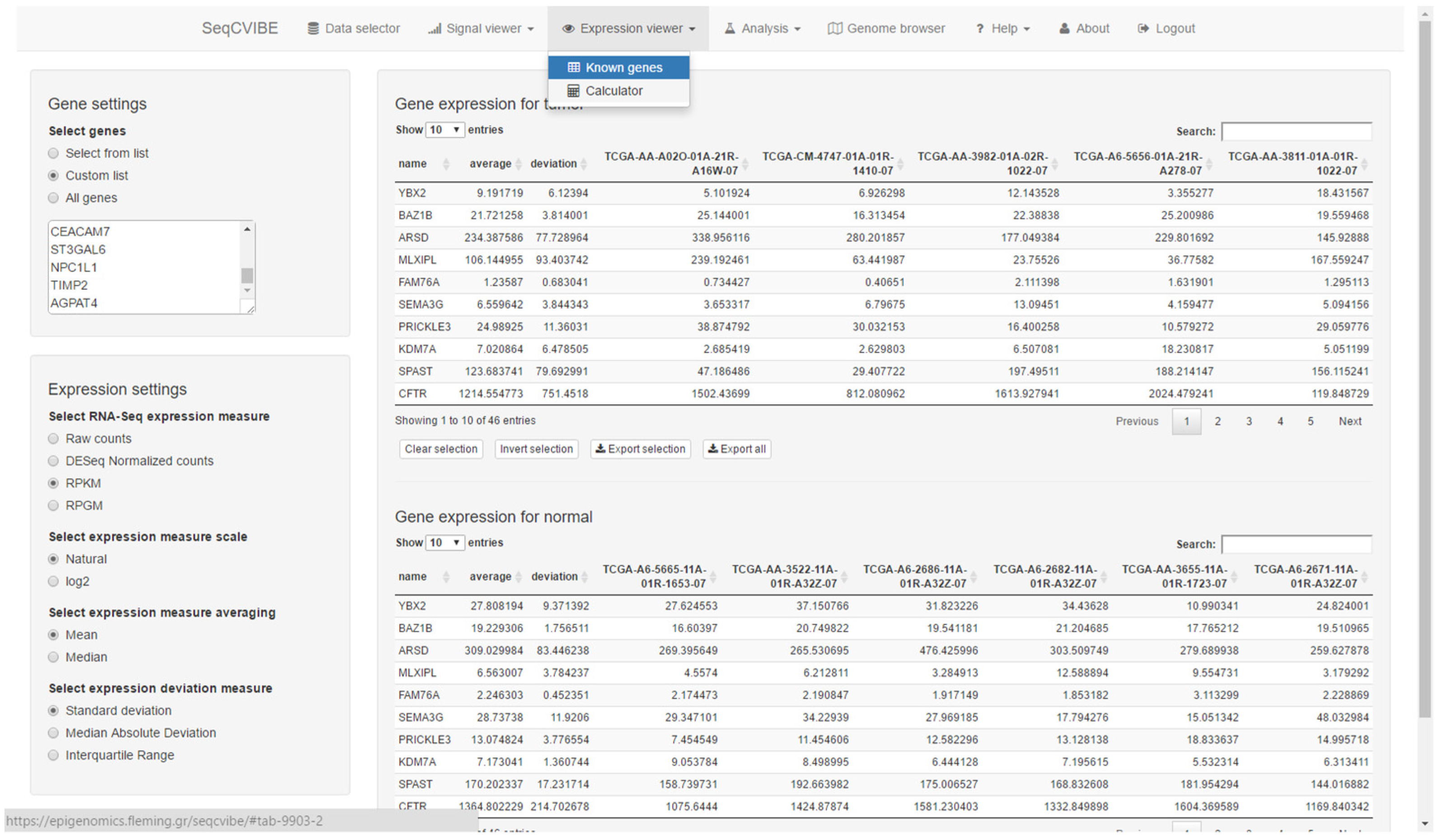
3.2.3. RNA Abundance Calculations
- Display gene expression values for known genes and annotated regions at various scales and summary metrics (raw, normalized, logarithmic, RPKM, etc.).
- Calculate abundances on-the-fly for non-annotated genomic regions. This functionality requires the presence of BAM files.
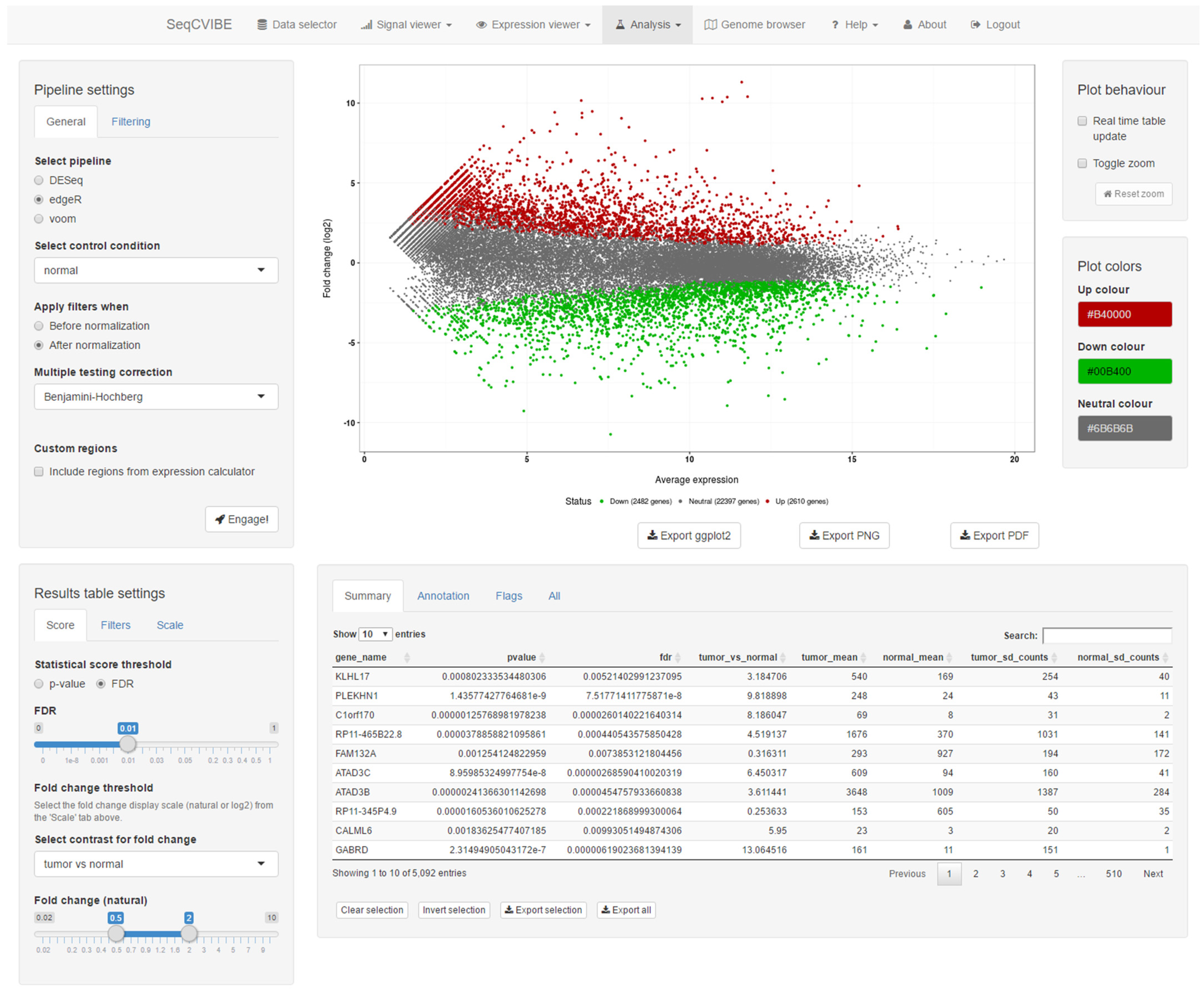
3.2.4. Real-Time DGEA and Exploration
- Joint analysis with several statistical methods available with metaseqR, reporting joint statistical outcomes.
- Visualization of the DGEA results with respect to statistical and differential abundance outcomes through an interactive MA plot which can be used to select and filter outcomes in real-time, along with displaying in table format.
- Various interactive filters for the selection of gene sets of interest, along with the display of gene expression values in multiple arithmetic scales.
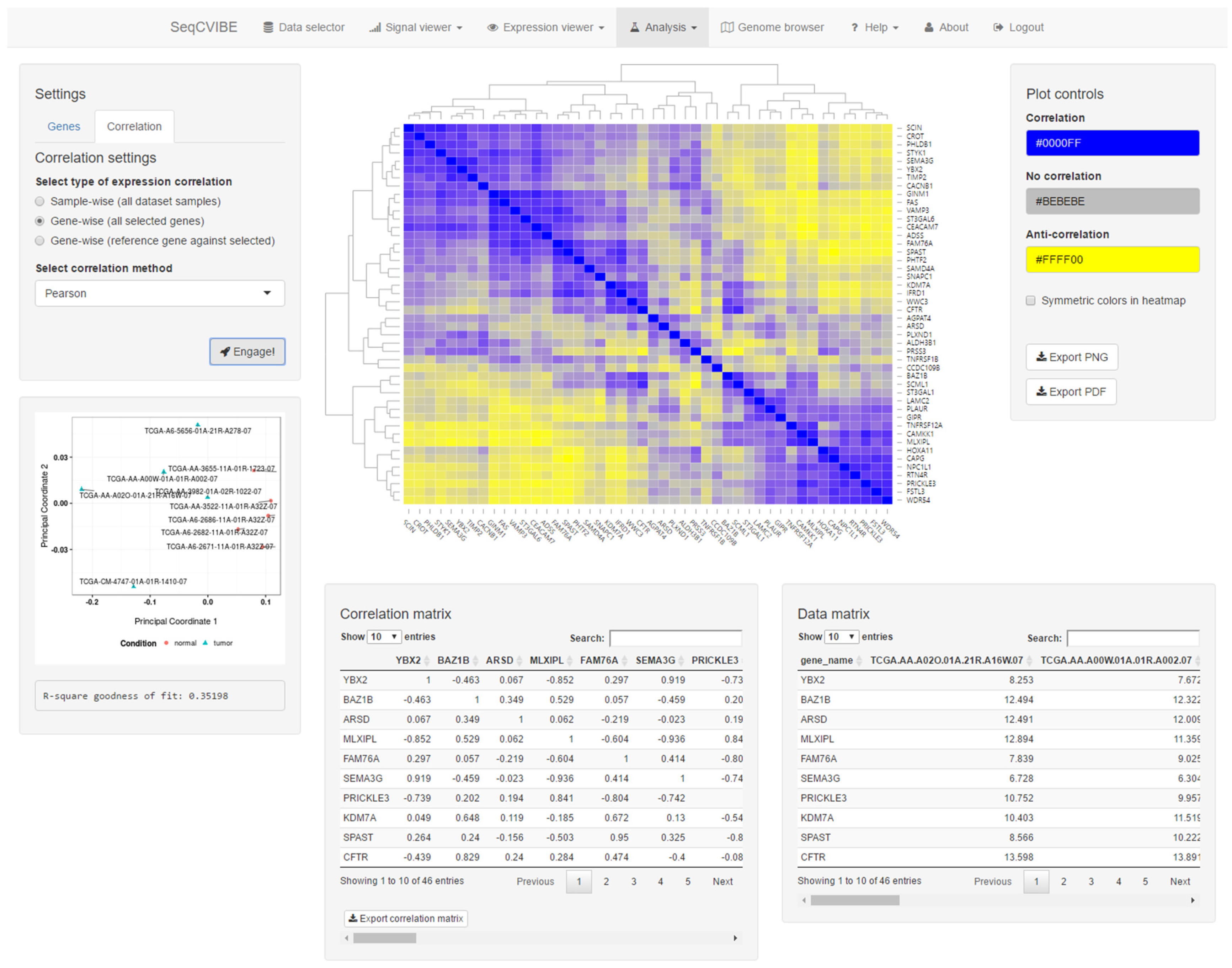
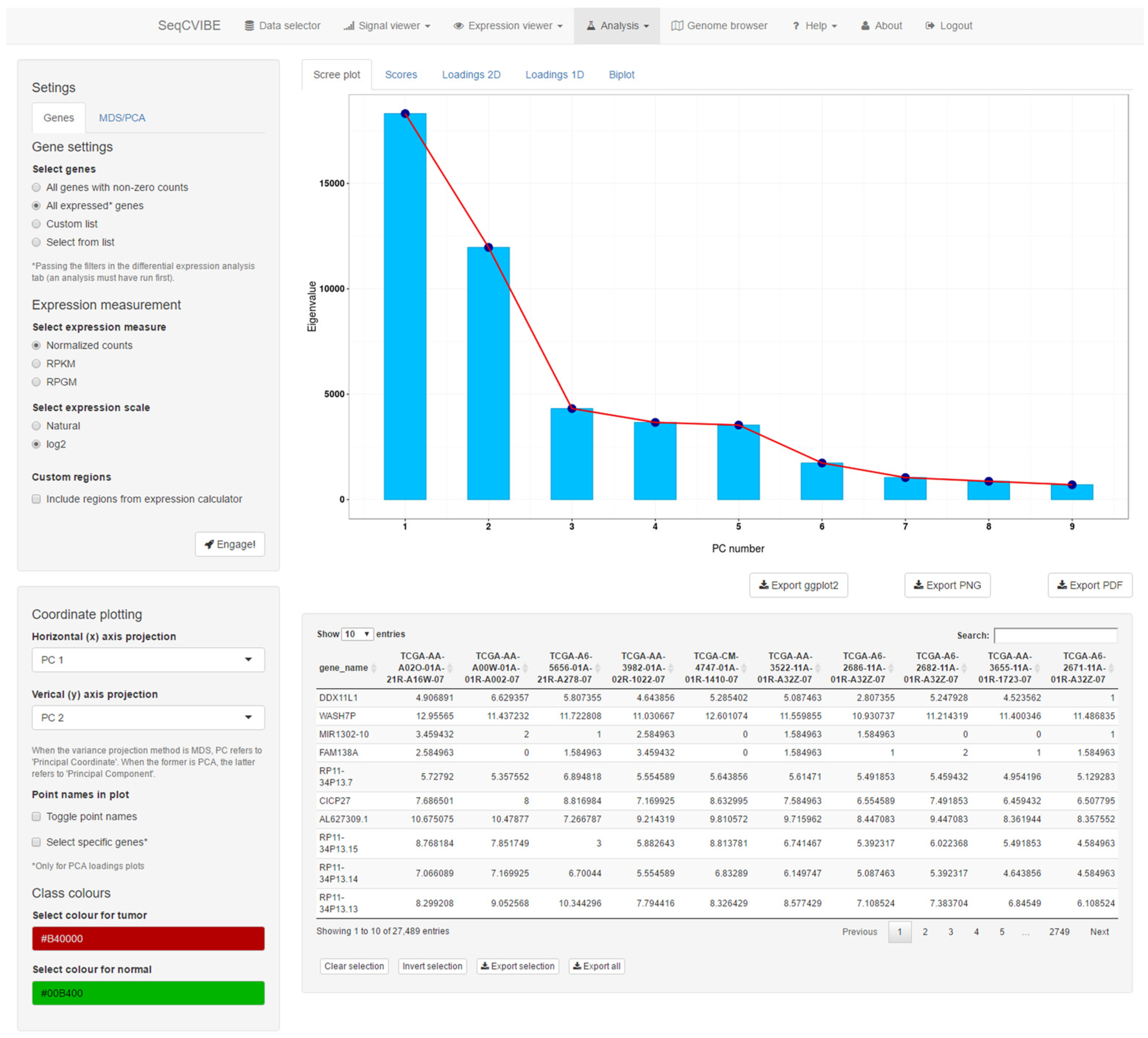
3.2.5. Additional Expression Analysis Tools
- Heatmap with the correlation values, also including hierarchical clustering for the estimation of sample similarities and the detection of potential outliers.
- Interactive tables with the correlation values and the expression values of the genes that are part of the correlation analysis.
- Real-time MDS plot using the genes and samples being analyzed, as additional means for estimating sample quality and outlier detection.
3.2.6. Genome Browsing
4. Results
4.1. Usage Scenarios
Usage scenario 1: My team has evidence on a novel long non-coding RNA with potential therapeutic properties in various cancer types. I want to check whether its expression affects nearby genes in my experimental as well as public data.
Usage scenario 2: My group studies liver development using the mouse as a model organism. I believe I have discovered a gene signature which significantly contributes in combination with the activity of known Transcription Factors (TFs) and I want to perform DGEA and clustering and correlation analysis. In addition, I want to check my gene signature on a mouse dataset studying brain development to check for potential correlations or conservation patterns.
Usage scenario 3: I teach Bioinformatics to Biology students and I want to prepare a workshop where I will demonstrate the basics of gene expression analysis using various examples of RNA-Seq data, as well as other generic aspects, such as genome browsing. I want a tool that can meet the requirements and also provide real-time analytics for discussion. Finally, as the workshop is addressed to bench biologists, I want to minimize the required computational skills.
Usage scenario 4: I am a Bioinformatics teaching assistant and I want to prepare a workshop addressed to math and/or computer science students without biological background. I will demonstrate the basics of gene expression analysis using various examples of RNA-Seq data but also explain biological signal and concepts such as exons. I want a tool that can meet the requirements, provide real-time analytics for discussion, and also provide intuitive visualizations for basic biological concepts regarding gene expression.
4.2. Public Deployment Example of SeqCVIBE and Datasets Hosted
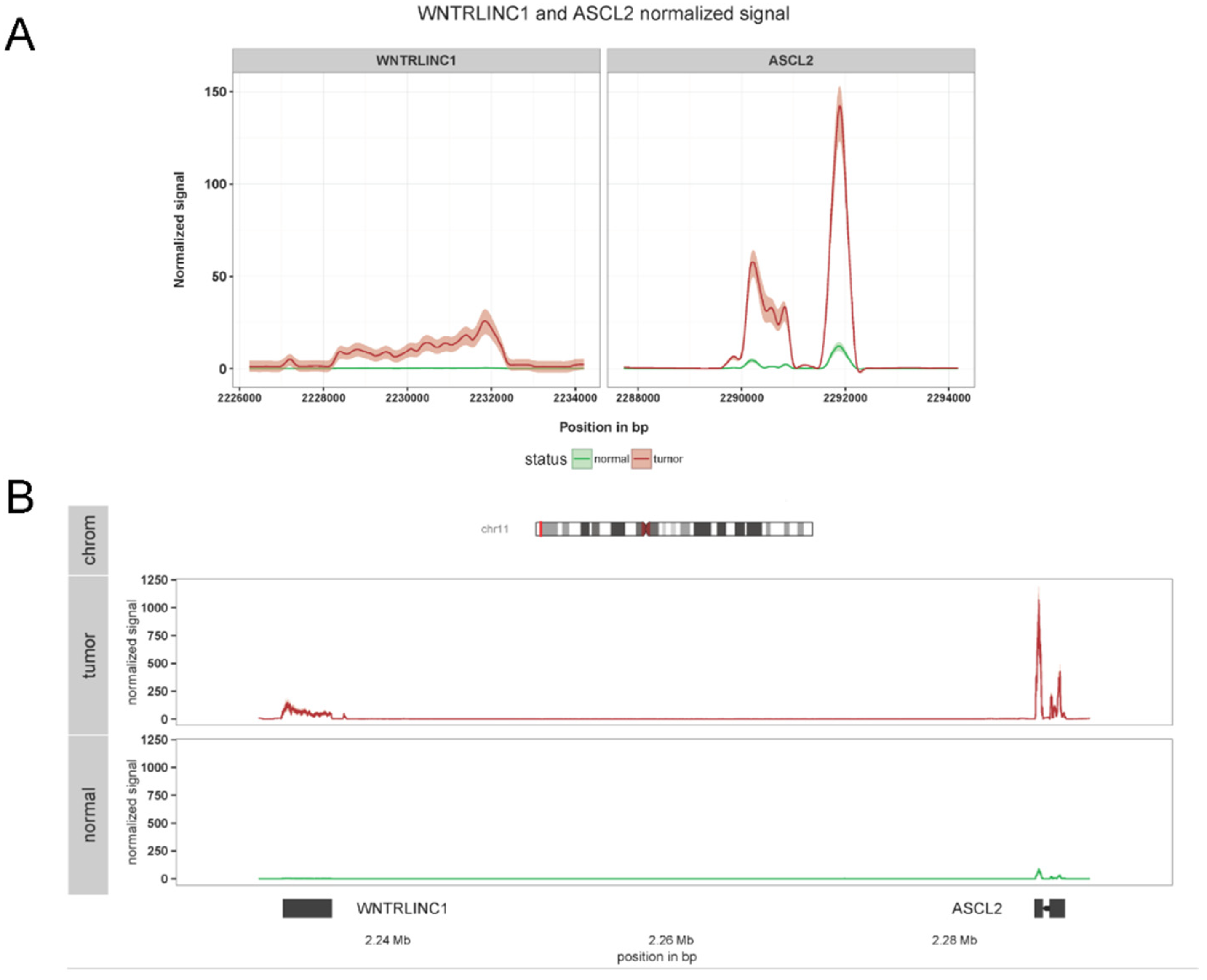
4.3. Investigation of the Role of WiNTRLINC1 in Wnt Signaling
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hölzer, M.; Marz, M. De Novo Transcriptome Assembly: A Comprehensive Cross-Species Comparison of Short-Read RNA-Seq Assemblers. GigaScience 2019, 8, giz039. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Hu, J.; Xue, C.; Zhang, H.; Susztak, K.; Reilly, M.P.; Xiao, R.; Li, M. ASEP: Gene-Based Detection of Allele-Specific Expression across Individuals in a Population by RNA Sequencing. PLoS Genet. 2020, 16, e1008786. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.R.; Tang, X.; Schultz-Rogers, L.E.; Vidal-Folch, N.; Jenkinson, W.G.; Schwab, T.L.; Gaonkar, K.; Cousin, M.A.; Nair, A.; Basu, S.; et al. A Tailored Approach to Fusion Transcript Identification Increases Diagnosis of Rare Inherited Disease. PLoS ONE 2019, 14, e0223337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Gao, S.; Muegge, K.; Zhang, W.; Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform. Biol. Insights 2015, 9s1, BBI.S28991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, A.P.; Poulain, S.; Fujii, H.; Miranda-Saavedra, D. Discovery and Characterization of New Transcripts from RNA-Seq Data in Mouse CD4+ T Cells. Genomics 2012, 100, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Kumar, V.; Jan, A.T.; Asger, M. Single-Cell Transcriptomics: Current Methods and Challenges in Data Acquisition and Analysis. Front. Neurosci. 2021, 15, 398. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isser, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g: Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Fanidis, D.; Moulos, P. Integrative, Normalization-Insusceptible Statistical Analysis of RNA-Seq Data, with Improved Differential Expression and Unbiased Downstream Functional Analysis. Brief. Bioinform. 2021, 22, bbaa156. [Google Scholar] [CrossRef]
- Assefa, A.T.; De Paepe, K.; Everaert, C.; Mestdagh, P.; Thas, O.; Vandesompele, J. Differential Gene Expression Analysis Tools Exhibit Substandard Performance for Long Non-Coding RNA-Sequencing Data. Genome Biol. 2018, 19, 96. [Google Scholar] [CrossRef] [Green Version]
- Moulos, P.; Hatzis, P. Systematic Integration of RNA-Seq Statistical Algorithms for Accurate Detection of Differential Gene Expression Patterns. Nucleic Acids Res. 2015, 43, e25. [Google Scholar] [CrossRef]
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F.; Parker, J.S.; Reynolds, S.M.; Silva, T.C.; Wang, L.-B.; Zhou, W.; Akbani, R.; Bailey, M.; Balu, S.; Berman, B.P.; et al. Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons’ Data. Cell Syst. 2019, 9, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. The Big Challenges of Big Data. Nature 2013, 498, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; Cook, D.; Lawrence, M. Ggbio: An R Package for Extending the Grammar of Graphics for Genomic Data. Genome Biol. 2012, 13, R77. [Google Scholar] [CrossRef] [Green Version]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A Dynamic Web Platform for Genome Visualization and Analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/Beta-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal. Cancer. Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Giakountis, A.; Moulos, P.; Zarkou, V.; Oikonomou, C.; Harokopos, V.; Hatzigeorgiou, A.G.; Reczko, M.; Hatzis, P. A Positive Regulatory Loop between a Wnt-Regulated Non-Coding RNA and ASCL2 Controls Intestinal Stem Cell Fate. Cell Rep. 2016, 15, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; van den Born, M.; Guryev, V.; Oving, I.; et al. Transcription Factor Achaete Scute-like 2 Controls Intestinal Stem Cell Fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Venco, F.; Vaskin, Y.; Ceol, A.; Muller, H. SMITH: A LIMS for Handling next-Generation Sequencing Workflows. BMC Bioinform. 2014, 15, S3. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.K.; Piehler, B.; Eckels, J.; Rauch, A.; Bellew, M.; Hussey, P.; Ramsay, S.; Nathe, C.; Lum, K.; Krouse, K.; et al. LabKey Server: An Open Source Platform for Scientific Data Integration, Analysis and Collaboration. BMC Bioinform. 2011, 12, 71. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.J. The Harmonic Mean P-Value for Combining Dependent Tests. Proc. Natl. Acad. Sci. USA 2019, 116, 1195–1200. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. Ngs.Plot: Quick Mining and Visualization of next-Generation Sequencing Data by Integrating Genomic Databases. BMC Genom. 2014, 15, 284. [Google Scholar] [CrossRef] [Green Version]
- Moulos, P. Recoup: Flexible and Versatile Signal Visualization from next Generation Sequencing. BMC Bioinform. 2021, 22, 2. [Google Scholar] [CrossRef]
- Fanidis, D.; Moulos, P.; Aidinis, V. Fibromine Is a Multi-Omics Database and Mining Tool for Target Discovery in Pulmonary Fibrosis. Sci. Rep. 2021, 11, 21712. [Google Scholar] [CrossRef]
- Klein, J.; Jupp, S.; Moulos, P.; Fernandez, M.; Buffin-Meyer, B.; Casemayou, A.; Chaaya, R.; Charonis, A.; Bascands, J.-L.; Stevens, R.; et al. The KUPKB: A Novel Web Application to Access Multiomics Data on Kidney Disease. FASEB J. 2012, 26, 2145–2153. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, C.; Qi, R.; Fu, H.; Ma, Q. ScREAD: A Single-Cell RNA-Seq Database for Alzheimer’s Disease. iScience 2020, 23, 101769. [Google Scholar] [CrossRef]
- Robinson, A.J.; Tamiru, M.; Salby, R.; Bolitho, C.; Williams, A.; Huggard, S.; Fisch, E.; Unsworth, K.; Whelan, J.; Lewsey, M.G. AgriSeqDB: An Online RNA-Seq Database for Functional Studies of Agriculturally Relevant Plant Species. BMC Plant Biol. 2018, 18, 200. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bothos, E.; Hatzis, P.; Moulos, P. Interactive Analysis, Exploration, and Visualization of RNA-Seq Data with SeqCVIBE. Methods Protoc. 2022, 5, 27. https://doi.org/10.3390/mps5020027
Bothos E, Hatzis P, Moulos P. Interactive Analysis, Exploration, and Visualization of RNA-Seq Data with SeqCVIBE. Methods and Protocols. 2022; 5(2):27. https://doi.org/10.3390/mps5020027
Chicago/Turabian StyleBothos, Efthimios, Pantelis Hatzis, and Panagiotis Moulos. 2022. "Interactive Analysis, Exploration, and Visualization of RNA-Seq Data with SeqCVIBE" Methods and Protocols 5, no. 2: 27. https://doi.org/10.3390/mps5020027
APA StyleBothos, E., Hatzis, P., & Moulos, P. (2022). Interactive Analysis, Exploration, and Visualization of RNA-Seq Data with SeqCVIBE. Methods and Protocols, 5(2), 27. https://doi.org/10.3390/mps5020027