
Tn5 DNA Transposase in Multi-Omics Research


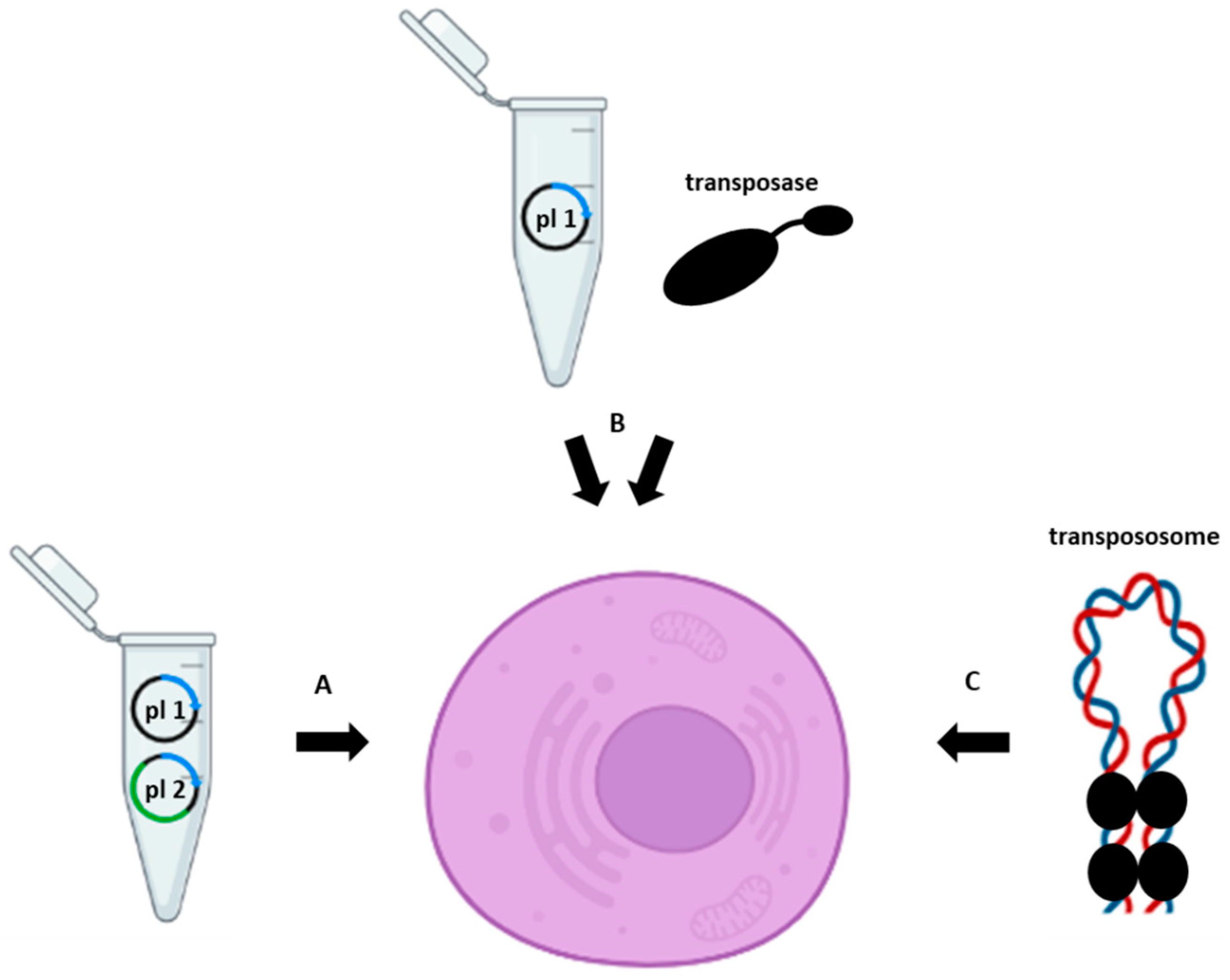{kind=link}
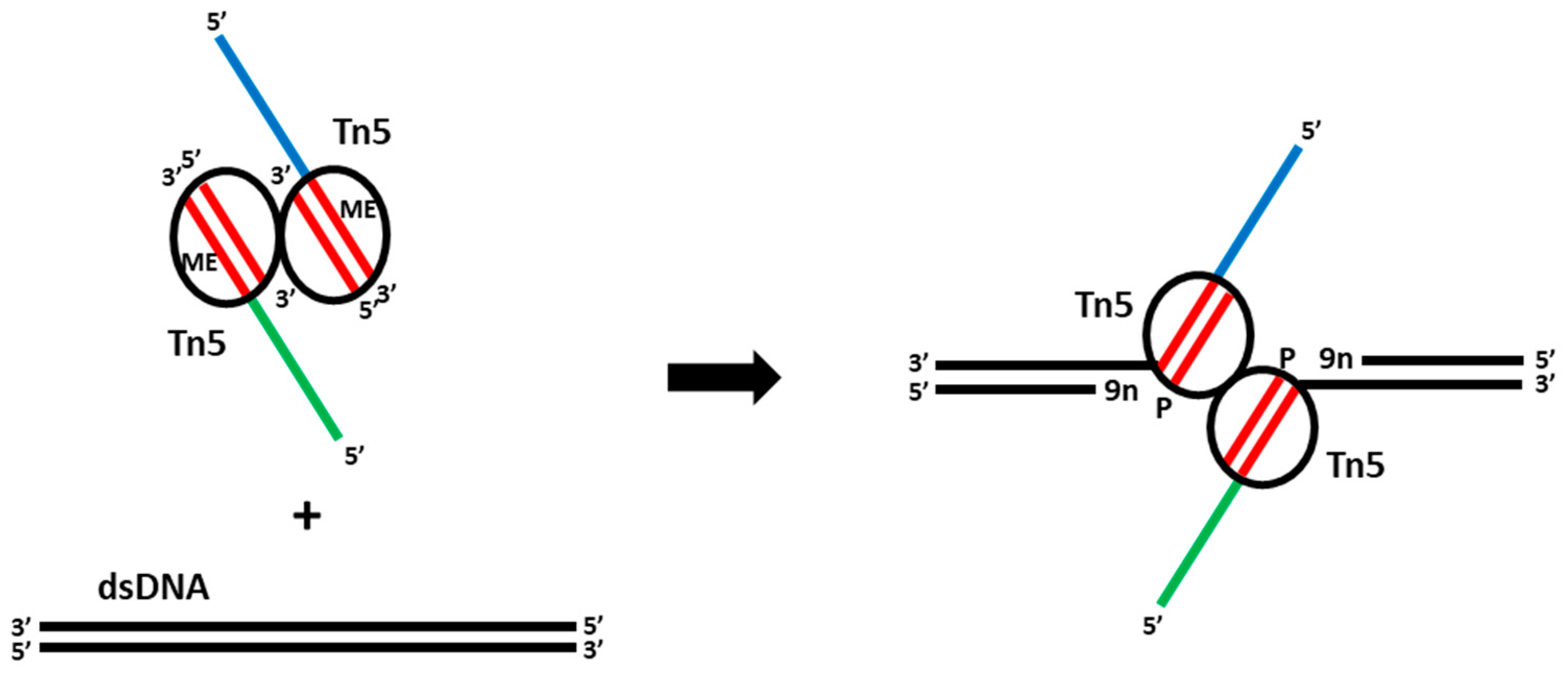{kind=link}
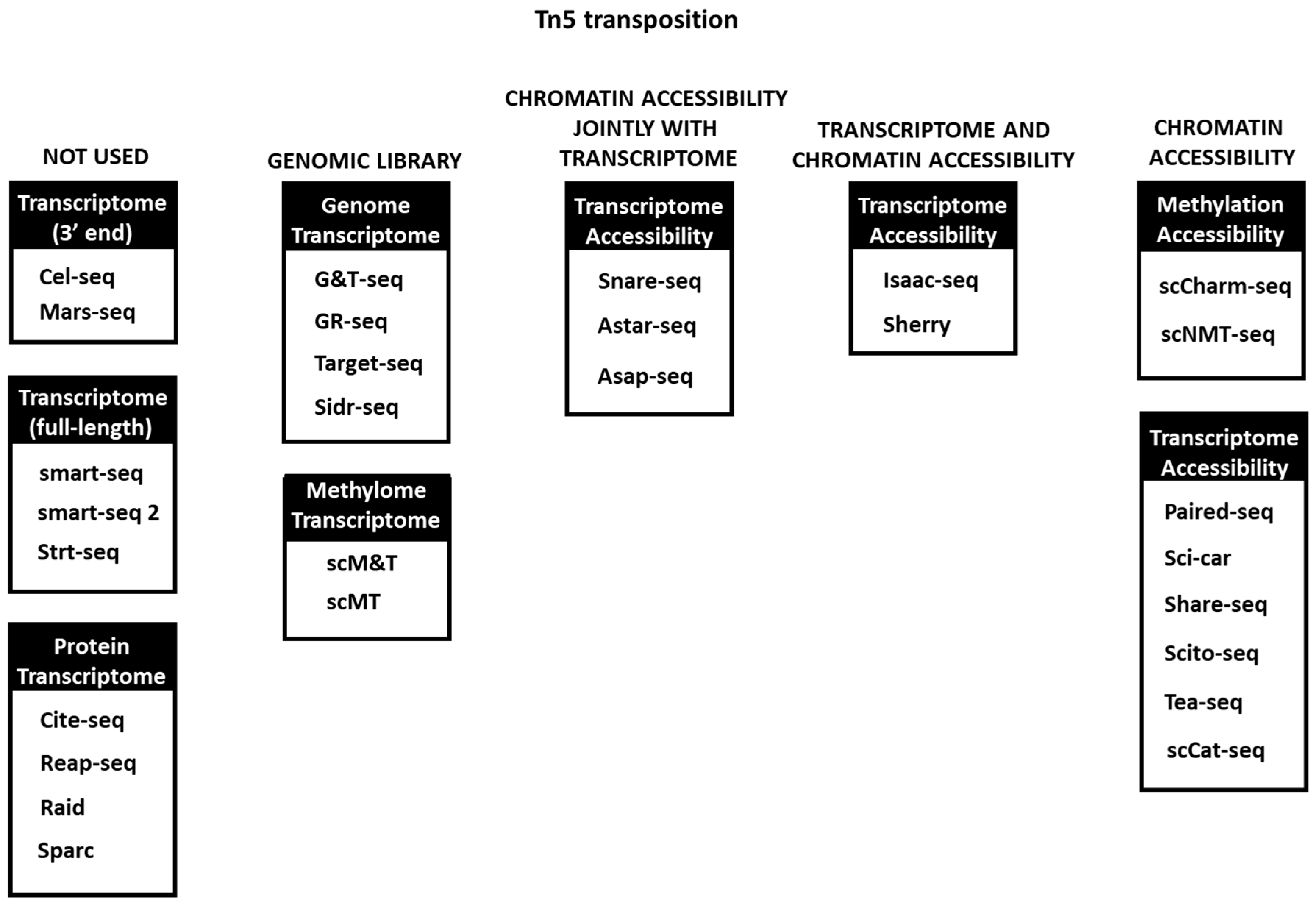{kind=link}
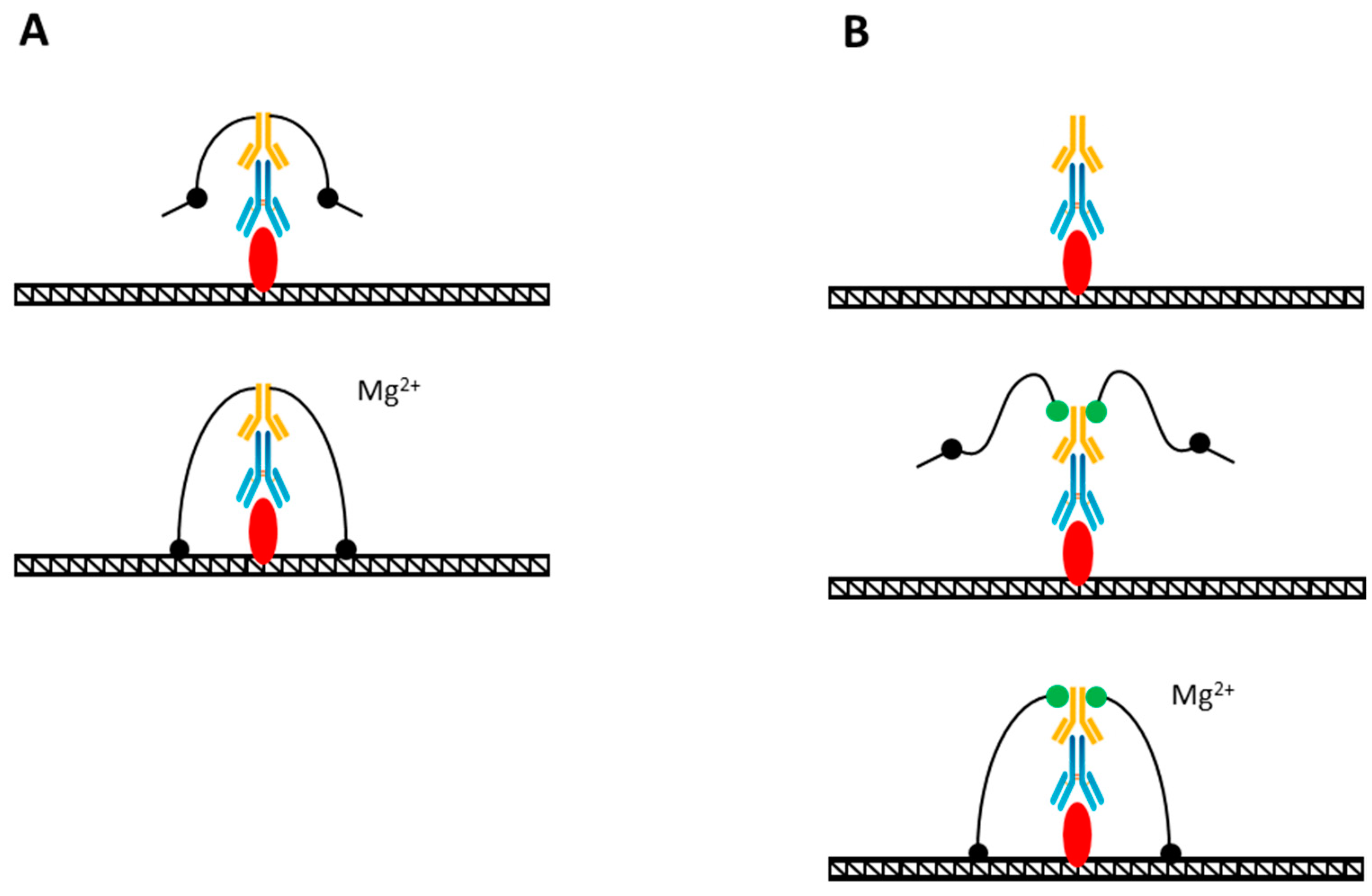{kind=link}
{kind=link}
Abstract
:1. An Overview of DNA Transposases
2. Tn5-Assisted Tagmentation
3. Tn5 Tagmentation in Single Cell Omics Technologies
4. Tn5 Tagmentation in Genome-Wide Profiling Assays
5. Tn5-Hybrid Tagmentation
6. Future Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. The Origin and Behavior of Mutable Loci in Maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Mutable Loci in Maize. Available online: https://profiles.nlm.nih.gov/spotlight/ll/catalog/nlm:nlmuid-101584613X32-doc (accessed on 10 February 2023).
- Hayward, A.; Gilbert, C. Transposable Elements. Curr. Biol. 2022, 32, R904–R909. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tao, X.; Yuan, S.; Zhang, Y.; Li, P.; Beilinson, H.A.; Zhang, Y.; Yu, W.; Pontarotti, P.; Escriva, H.; et al. Discovery of an Active RAG Transposon Illuminates the Origins of V(D)J Recombination. Cell 2016, 166, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Hickman, A.B.; Dyda, F. DNA Transposition at Work. Chem. Rev. 2016, 116, 12758–12784. [Google Scholar] [CrossRef] [PubMed]
- Nesmelova, I.V.; Hackett, P.B. DDE Transposases: Structural Similarity and Diversity. Adv. Drug Deliv. Rev. 2010, 62, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaño, S.P.; Pigli, Y.Z.; Rice, P.A. Structure of the Mu Transpososome Illuminates Evolution of DDE Recombinases. Nature 2012, 491, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Gogol-Döring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvák, Z.; Ivics, Z. Genome-Wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the PiggyBac Transposon in Primary Human CD4+ T Cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, R.; Kokubu, C.; Yusa, K.; Keng, V.W.; Horie, K.; Takeda, J. Sleeping Beauty Transposase Has an Affinity for Heterochromatin Conformation. Mol. Cell. Biol. 2007, 27, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Savilahti, H.; Mizuuchi, K. Mu Transpositional Recombination: Donor DNA Cleavage and Strand Transfer in Trans by the Mu Transposase. Cell 1996, 85, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Maiti, S.; Huls, H.; Singh, H.; Olivares, S.; Mátés, L.; Izsvák, Z.; Ivics, Z.; Lee, D.A.; Champlin, R.E.; et al. The Hyperactive Sleeping Beauty Transposase SB100X Improves the Genetic Modification of T Cells to Express a Chimeric Antigen Receptor. Gene Ther. 2011, 18, 849–856. [Google Scholar] [CrossRef]
- Rasila, T.S.; Pulkkinen, E.; Kiljunen, S.; Haapa-Paananen, S.; Pajunen, M.I.; Salminen, A.; Paulin, L.; Vihinen, M.; Rice, P.A.; Savilahti, H. Mu Transpososome Activity-Profiling Yields Hyperactive MuA Variants for Highly Efficient Genetic and Genome Engineering. Nucleic Acids Res. 2018, 46, 4649–4661. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Ivics, Z. Non-Viral Therapeutic Cell Engineering with the Sleeping Beauty Transposon System. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval-Villegas, N.; Nurieva, W.; Amberger, M.; Ivics, Z. Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, PiggyBac and Tol2 for Genome Engineering. Int. J. Mol. Sci. 2021, 22, 5084. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Izsvák, Z.; Narayanavari, S.A.; Singh, H.; Ivics, Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet. 2017, 33, 852–870. [Google Scholar] [CrossRef]
- Goryshin, I.Y.; Jendrisak, J.; Hoffman, L.M.; Meis, R.; Reznikoff, W.S. Insertional Transposon Mutagenesis by Electroporation of Released Tn5 Transposition Complexes. Nat. Biotechnol. 2000, 18, 97–100. [Google Scholar] [CrossRef]
- Akhverdyan, V.Z.; Gak, E.R.; Tokmakova, I.L.; Stoynova, N.V.; Yomantas, Y.A.V.; Mashko, S.V. Application of the Bacteriophage Mu-Driven System for the Integration/Amplification of Target Genes in the Chromosomes of Engineered Gram-Negative Bacteria—Mini Review. Appl. Microbiol. Biotechnol. 2011, 91, 857–871. [Google Scholar] [CrossRef] [Green Version]
- Lamberg, A.; Nieminen, S.; Qiao, M.; Savilahti, H. Efficient Insertion Mutagenesis Strategy for Bacterial Genomes Involving Electroporation of In Vitro-Assembled DNA Transposition Complexes of Bacteriophage Mu. Appl. Environ. Microbiol. 2002, 68, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Pajunen, M.I.; Pulliainen, A.T.; Finne, J.; Savilahti, H. Generation of Transposon Insertion Mutant Libraries for Gram-Positive Bacteria by Electroporation of Phage Mu DNA Transposition Complexes. Microbiology 2005, 151, 1209–1218. [Google Scholar] [CrossRef] [Green Version]
- Paatero, A.O.; Turakainen, H.; Happonen, L.J.; Olsson, C.; Palomäki, T.; Pajunen, M.I.; Meng, X.; Otonkoski, T.; Tuuri, T.; Berry, C.; et al. Bacteriophage Mu Integration in Yeast and Mammalian Genomes. Nucleic Acids Res. 2008, 36, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S. Single-Cell RNA-Sequencing: The Future of Genome Biology Is Now. RNA Biol. 2017, 14, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Björklund, Å.K.; Reinius, B.; Sagasser, S.; Winberg, G.; Sandberg, R. Tn5 Transposase and Tagmentation Procedures for Massively Scaled Sequencing Projects. Genome Res. 2014, 24, 2033–2040. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Jin, K.; Bai, Y.; Fu, H.; Liu, L.; Liu, B. Tn5 Transposase Applied in Genomics Research. Int. J. Mol. Sci. 2020, 21, 8329. [Google Scholar] [CrossRef]
- Wei, M.; Mi, C.-L.; Jing, C.-Q.; Wang, T.-Y. Progress of Transposon Vector System for Production of Recombinant Therapeutic Proteins in Mammalian Cells. Front. Bioeng. Biotechnol. 2022, 10, 879222. [Google Scholar] [CrossRef]
- Cain, A.K.; Barquist, L.; Goodman, A.L.; Paulsen, I.T.; Parkhill, J.; van Opijnen, T. A Decade of Advances in Transposon-Insertion Sequencing. Nat. Rev. Genet. 2020, 21, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, K. In Vitro Transposition of Bacteriophage Mu: A Biochemical Approach to a Novel Replication Reaction. Cell 1983, 35, 785–794. [Google Scholar] [CrossRef]
- Savilahti, H.; Rice, P.A.; Mizuuchi, K. The Phage Mu Transpososome Core: DNA Requirements for Assembly and Function. EMBO J. 1995, 14, 4893–4903. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21.29.1–21.29.9. [Google Scholar] [CrossRef] [Green Version]
- Nature Protocols. Chromatin Accessibility Profiling by ATAC-Seq. Available online: https://www.nature.com/articles/s41596-022-00692-9 (accessed on 7 October 2022).
- Adey, A.; Morrison, H.G.; Asan; Xun, X.; Kitzman, J.O.; Turner, E.H.; Stackhouse, B.; MacKenzie, A.P.; Caruccio, N.C.; Zhang, X.; et al. Rapid, Low-Input, Low-Bias Construction of Shotgun Fragment Libraries by High-Density in Vitro Transposition. Genome Biol. 2010, 11, R119. [Google Scholar] [CrossRef] [Green Version]
- MuA Transposase. Domus Biotechnologies. Available online: https://domusbiotechnologies.com (accessed on 7 October 2022).
- Naumann, T.A.; Reznikoff, W.S. Tn 5 Transposase Active Site Mutants. J. Biol. Chem. 2002, 277, 17623–17629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinov, G.K.; Williams, B.A.; McCue, K.; Schroth, G.P.; Gertz, J.; Myers, R.M.; Wold, B.J. From Single-Cell to Cell-Pool Transcriptomes: Stochasticity in Gene Expression and RNA Splicing. Genome Res. 2014, 24, 496–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trombetta, J.J.; Gennert, D.; Lu, D.; Satija, R.; Shalek, A.K.; Regev, A. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr. Protoc. Mol. Biol. 2014, 107, 4.22.1–4.22.17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-Length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; de Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive Highly-Multiplexed Single-Cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Yanai, I.; Hashimshony, T. CEL-Seq2-Single-Cell RNA Sequencing by Multiplexed Linear Amplification. Methods Mol. Biol. 2019, 1979, 45–56. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Kenigsberg, E.; Jaitin, D.A.; David, E.; Paul, F.; Tanay, A.; Amit, I. MARS-Seq2.0: An Experimental and Analytical Pipeline for Indexed Sorting Combined with Single-Cell RNA Sequencing. Nat. Protoc. 2019, 14, 1841–1862. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, K.N. Single-Cell Tagged Reverse Transcription (STRT-Seq). In Single Cell Methods: Sequencing and Proteomics; Proserpio, V., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 133–153. ISBN 978-1-4939-9240-9. [Google Scholar]
- Hochgerner, H.; Lönnerberg, P.; Hodge, R.; Mikes, J.; Heskol, A.; Hubschle, H.; Lin, P.; Picelli, S.; La Manno, G.; Ratz, M.; et al. STRT-Seq-2i: Dual-Index 5ʹ Single Cell and Nucleus RNA-Seq on an Addressable Microwell Array. Sci. Rep. 2017, 7, 16327. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-Wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively Parallel Digital Transcriptional Profiling of Single Cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Ogbeide, S.; Giannese, F.; Mincarelli, L.; Macaulay, I.C. Into the Multiverse: Advances in Single-Cell Multiomic Profiling. Trends Genet. 2022, 38, 831–843. [Google Scholar] [CrossRef]
- Dey, S.S.; Kester, L.; Spanjaard, B.; Bienko, M.; van Oudenaarden, A. Integrated Genome and Transcriptome Sequencing of the Same Cell. Nat. Biotechnol. 2015, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Meira, A.; O’Sullivan, J.; Rahman, H.; Mead, A.J. TARGET-Seq: A Protocol for High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. STAR Protoc. 2020, 1, 100125. [Google Scholar] [CrossRef] [PubMed]
- Han, K.Y.; Kim, K.-T.; Joung, J.-G.; Son, D.-S.; Kim, Y.J.; Jo, A.; Jeon, H.-J.; Moon, H.-S.; Yoo, C.E.; Chung, W.; et al. SIDR: Simultaneous Isolation and Parallel Sequencing of Genomic DNA and Total RNA from Single Cells. Genome Res. 2018, 28, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, I.C.; Haerty, W.; Kumar, P.; Li, Y.I.; Hu, T.X.; Teng, M.J.; Goolam, M.; Saurat, N.; Coupland, P.; Shirley, L.M.; et al. G&T-Seq: Parallel Sequencing of Single-Cell Genomes and Transcriptomes. Nat. Methods 2015, 12, 519–522. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Huang, K.; An, Q.; Du, G.; Hu, G.; Xue, J.; Zhu, X.; Wang, C.-Y.; Xue, Z.; Fan, G. Simultaneous Profiling of Transcriptome and DNA Methylome from a Single Cell. Genome Biol. 2016, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.A.; Ponting, C.P.; Voet, T.; et al. Parallel Single-Cell Sequencing Links Transcriptional and Epigenetic Heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, C.; Quintero, A.; Wu, L.; Yuan, Y.; Wang, M.; Cheng, M.; Leng, L.; Xu, L.; Dong, G.; et al. Deconvolution of Single-Cell Multi-Omics Layers Reveals Regulatory Heterogeneity. Nat. Commun. 2019, 10, 470. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint Profiling of Chromatin Accessibility and Gene Expression in Thousands of Single Cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Yu, M.; Huang, H.; Juric, I.; Abnousi, A.; Hu, R.; Lucero, J.; Behrens, M.M.; Hu, M.; Ren, B. An Ultra High-Throughput Method for Single-Cell Joint Analysis of Open Chromatin and Transcriptome. Nat. Struct. Mol. Biol. 2019, 26, 1063–1070. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; LaFave, L.M.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116.e20. [Google Scholar] [CrossRef]
- Swanson, E.; Lord, C.; Reading, J.; Heubeck, A.T.; Genge, P.C.; Thomson, Z.; Weiss, M.D.; Li, X.; Savage, A.K.; Green, R.R.; et al. Simultaneous Trimodal Single-Cell Measurement of Transcripts, Epitopes, and Chromatin Accessibility Using TEA-Seq. eLife 2021, 10, e63632. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, D.S.; Tamaki, W.; Sun, Y.; Ogorodnikov, A.; Hartoularos, G.C.; Winters, A.; Yeung, B.Z.; Nazor, K.L.; Song, Y.S.; et al. SCITO-Seq: Single-Cell Combinatorial Indexed Cytometry Sequencing. Nat. Methods 2021, 18, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Cheng, X.; Guo, F. Protocol for ScChaRM-Seq: Simultaneous Profiling of Gene Expression, DNA Methylation, and Chromatin Accessibility in Single Cells. STAR Protoc. 2021, 2, 100972. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Gu, C.; You, D.; Huang, Z.; Qian, J.; Yang, Q.; Cheng, X.; Zhang, L.; Wang, H.; Wang, P.; et al. Decoding Dynamic Epigenetic Landscapes in Human Oocytes Using Single-Cell Multi-Omics Sequencing. Cell Stem Cell 2021, 28, 1641–1656.e7. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.-A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. ScNMT-Seq Enables Joint Profiling of Chromatin Accessibility DNA Methylation and Transcription in Single Cells. Nat. Commun. 2018, 9, 781. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lake, B.B.; Zhang, K. High-Throughput Sequencing of the Transcriptome and Chromatin Accessibility in the Same Cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.R.; Farran, C.A.E.; Zeng, Y.Y.; Yi, Y.; Warrier, T.; Gautam, P.; Collins, J.J.; Xu, J.; Dröge, P.; Koh, C.-G.; et al. Parallel Bimodal Single-Cell Sequencing of Transcriptome and Chromatin Accessibility. Genome Res. 2020, 30, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, Multimodal Profiling of Chromatin Accessibility, Gene Expression and Protein Levels in Single Cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, W.; Zhang, Y.; Chen, Y.; Hong, N.; Zhang, Q.; Wang, X.; Hu, Y.; Song, K.; Jin, W.; et al. ISSAAC-Seq Enables Sensitive and Flexible Multimodal Profiling of Chromatin Accessibility and Gene Expression in Single Cells. Nat. Methods 2022, 19, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Fu, Y.; Sun, Y.; Li, J.; Liu, L.; Yao, J.; Wang, G.; Wu, Y.; Lao, K.; Lee, R.W.; et al. RNA Sequencing by Direct Tagmentation of RNA/DNA Hybrids. Proc. Natl. Acad. Sci. USA 2020, 117, 2886–2893. [Google Scholar] [CrossRef] [Green Version]
- Visa, N.; Jordán-Pla, A. ChIP and ChIP-Related Techniques: Expanding the Fields of Application and Improving ChIP Performance. In Chromatin Immunoprecipitation: Methods and Protocols; Visa, N., Jordán-Pla, A., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; pp. 1–7. ISBN 978-1-4939-7380-4. [Google Scholar]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-Seq Guidelines and Practices of the ENCODE and ModENCODE Consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [Green Version]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xiong, H.; Ai, S.; Yu, X.; Liu, Y.; Zhang, J.; He, A. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 2019, 76, 206–216.e7. [Google Scholar] [CrossRef]
- Harada, A.; Maehara, K.; Handa, T.; Arimura, Y.; Nogami, J.; Hayashi-Takanaka, Y.; Shirahige, K.; Kurumizaka, H.; Kimura, H.; Ohkawa, Y. A Chromatin Integration Labelling Method Enables Epigenomic Profiling with Lower Input. Nat. Cell Biol. 2019, 21, 287–296. [Google Scholar] [CrossRef]
- Carter, B.; Ku, W.L.; Kang, J.Y.; Hu, G.; Perrie, J.; Tang, Q.; Zhao, K. Mapping Histone Modifications in Low Cell Number and Single Cells Using Antibody-Guided Chromatin Tagmentation (ACT-Seq). Nat. Commun. 2019, 10, 3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.J.; Furlan, S.N.; Mihalas, A.B.; Kaya-Okur, H.S.; Feroze, A.H.; Emerson, S.N.; Zheng, Y.; Carson, K.; Cimino, P.J.; Keene, C.D.; et al. Single-Cell CUT&Tag Analysis of Chromatin Modifications in Differentiation and Tumor Progression. Nat. Biotechnol. 2021, 39, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Bartosovic, M.; Kabbe, M.; Castelo-Branco, G. Single-Cell CUT&Tag Profiles Histone Modifications and Transcription Factors in Complex Tissues. Nat. Biotechnol. 2021, 39, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J.G.; Kaya-Okur, H.S.; Ahmad, K. Efficient Chromatin Accessibility Mapping in Situ by Nucleosome-Tethered Tagmentation. eLife 2020, 9, e63274. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G.; Ahmad, K. Simplified Epigenome Profiling Using Antibody-Tethered Tagmentation. Bio-Protocol 2021, 11, e4043. [Google Scholar] [CrossRef]
- Janssens, D.H.; Otto, D.J.; Meers, M.P.; Setty, M.; Ahmad, K.; Henikoff, S. CUT&Tag2for1: A Modified Method for Simultaneous Profiling of the Accessible and Silenced Regulome in Single Cells. Genome Biol. 2022, 23, 81. [Google Scholar] [CrossRef]
- Bartlett, D.A.; Dileep, V.; Handa, T.; Ohkawa, Y.; Kimura, H.; Henikoff, S.; Gilbert, D.M. High-Throughput Single-Cell Epigenomic Profiling by Targeted Insertion of Promoters (TIP-Seq). J. Cell Biol. 2021, 220, e202103078. [Google Scholar] [CrossRef]
- Sati, S.; Jones, P.; Kim, H.S.; Zhou, L.A.; Rapp-Reyes, E.; Leung, T.H. HiCuT: An Efficient and Low Input Method to Identify Protein-Directed Chromatin Interactions. PLoS Genet. 2022, 18, e1010121. [Google Scholar] [CrossRef]
- Gopalan, S.; Wang, Y.; Harper, N.W.; Garber, M.; Fazzio, T.G. Simultaneous Profiling of Multiple Chromatin Proteins in the Same Cells. Mol. Cell 2021, 81, 4736–4746.e5. [Google Scholar] [CrossRef]
- Meers, M.P.; Llagas, G.; Janssens, D.H.; Codomo, C.A.; Henikoff, S. Multifactorial Profiling of Epigenetic Landscapes at Single-Cell Resolution Using MulTI-Tag. Nat. Biotechnol. 2022, 1–9. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in Situ Genome-Wide Profiling with High Efficiency for Low Cell Numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Meers, M.P.; Janssens, D.H.; Henikoff, S. Pioneer Factor-Nucleosome Binding Events during Differentiation Are Motif Encoded. Mol. Cell 2019, 75, 562–575.e5. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, M.; Giannese, F.; Lazarević, D.; Giansanti, V.; Rosano, D.; Monzani, S.; Catalano, I.; Grassi, E.; Zanella, E.R.; Botrugno, O.A.; et al. Chromatin Velocity Reveals Epigenetic Dynamics by Single-Cell Profiling of Heterochromatin and Euchromatin. Nat. Biotechnol. 2022, 40, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Mulqueen, R.M.; Pokholok, D.; O’Connell, B.L.; Thornton, C.A.; Zhang, F.; O’Roak, B.J.; Link, J.; Yardımcı, G.G.; Sears, R.C.; Steemers, F.J.; et al. High-Content Single-Cell Combinatorial Indexing. Nat. Biotechnol. 2021, 39, 1574–1580. [Google Scholar] [CrossRef]
- Miyanari, Y. Imaging Chromatin Accessibility by Assay of Transposase-Accessible Chromatin with Visualization. In Epigenomics: Methods and Protocols; Methods in Molecular Biology; Hatada, I., Horii, T., Eds.; Springer: New York, NY, USA, 2023; pp. 93–101. ISBN 978-1-07-162724-2. [Google Scholar]
- Moudgil, A.; Wilkinson, M.N.; Chen, X.; He, J.; Cammack, A.J.; Vasek, M.J.; Lagunas, T.; Qi, Z.; Lalli, M.A.; Guo, C.; et al. Self-Reporting Transposons Enable Simultaneous Readout of Gene Expression and Transcription Factor Binding in Single Cells. Cell 2020, 182, 992–1008.e21. [Google Scholar] [CrossRef]
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-Guided Retargeting of Sleeping Beauty Transposition in Human Cells. eLife 2020, 9, e53868. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penkov, D.; Zubkova, E.; Parfyonova, Y. Tn5 DNA Transposase in Multi-Omics Research. Methods Protoc. 2023, 6, 24. https://doi.org/10.3390/mps6020024
Penkov D, Zubkova E, Parfyonova Y. Tn5 DNA Transposase in Multi-Omics Research. Methods and Protocols. 2023; 6(2):24. https://doi.org/10.3390/mps6020024
Chicago/Turabian StylePenkov, Dmitry, Ekaterina Zubkova, and Yelena Parfyonova. 2023. "Tn5 DNA Transposase in Multi-Omics Research" Methods and Protocols 6, no. 2: 24. https://doi.org/10.3390/mps6020024
APA StylePenkov, D., Zubkova, E., & Parfyonova, Y. (2023). Tn5 DNA Transposase in Multi-Omics Research. Methods and Protocols, 6(2), 24. https://doi.org/10.3390/mps6020024