1. Introduction
Since ancient times, plants provide mankind with many remedies and food supplements, and in some countries, medicinal plants may be the primary or sole source of healthcare [
1,
2]. Some examples of medicinal and herbal plant products include chamomile, ephedra, garlic, ginseng, marijuana and opium. Furthermore, plants have a significant impact on diet in both societal (i.e., veganism) but also in practical contexts in relation to human physiology. The nutritional and medicinal benefits of plants are reflected in their phytochemical components, as plants are able to synthesise a large range of compounds known as phytochemicals [
3,
4,
5]. The metabolites of plants are categorised into two groups: primary and secondary metabolites. The primary metabolites are involved in the major metabolic pathways for plant growth and development, while secondary metabolites serve non-essential purposes in the plants [
6,
7]. Secondary metabolites are required for plants to survive as these compounds are involved in adaptation and survival mechanisms, but also have been proven to have medicinal properties [
4,
8]. As a result, medicinal and herbal plants play a critical role in the food and pharmaceutical industries [
9].
Several in vivo and in vitro studies have been focused on the beneficial biological actions of plant extracts highlighting their importance in ethnobotany. Most innovative studies emphasise the phytochemical characterisation and identification of biologically active compounds, which can then be synthesised in organic chemistry laboratories or in heterologous hosts [
10]. In this effort, several assays for the determination of the major categories of phytochemicals (i.e., polyphenols, tannins, flavonoids, alkaloids, etc.) were combined with assays focusing on the assessment of the antioxidant properties of plant extracts. These methods are useful for the identification of bioactive compounds. To date, there is not an optimised comprehensive protocol to provide a series of analytical options within a general (not detailed) phytochemical characterisation.
The present multiparametric protocol provides targeted assays for the characterisation of the main categories of plant metabolites for eight distinct biochemical protocols with standardised microplate assays. Specifically, flavonoids are quantified by their reaction with aluminium trichloride [
11], tannins are assayed by their reaction with vanillin under acidic conditions [
12], and polyphenols are assessed by the Folin reagent [
13]. In relation to the metal scavenging [
14] and the antioxidant potential [
15] of plant extracts, the capacity of plant extracts to reduce ions such as iron and copper (
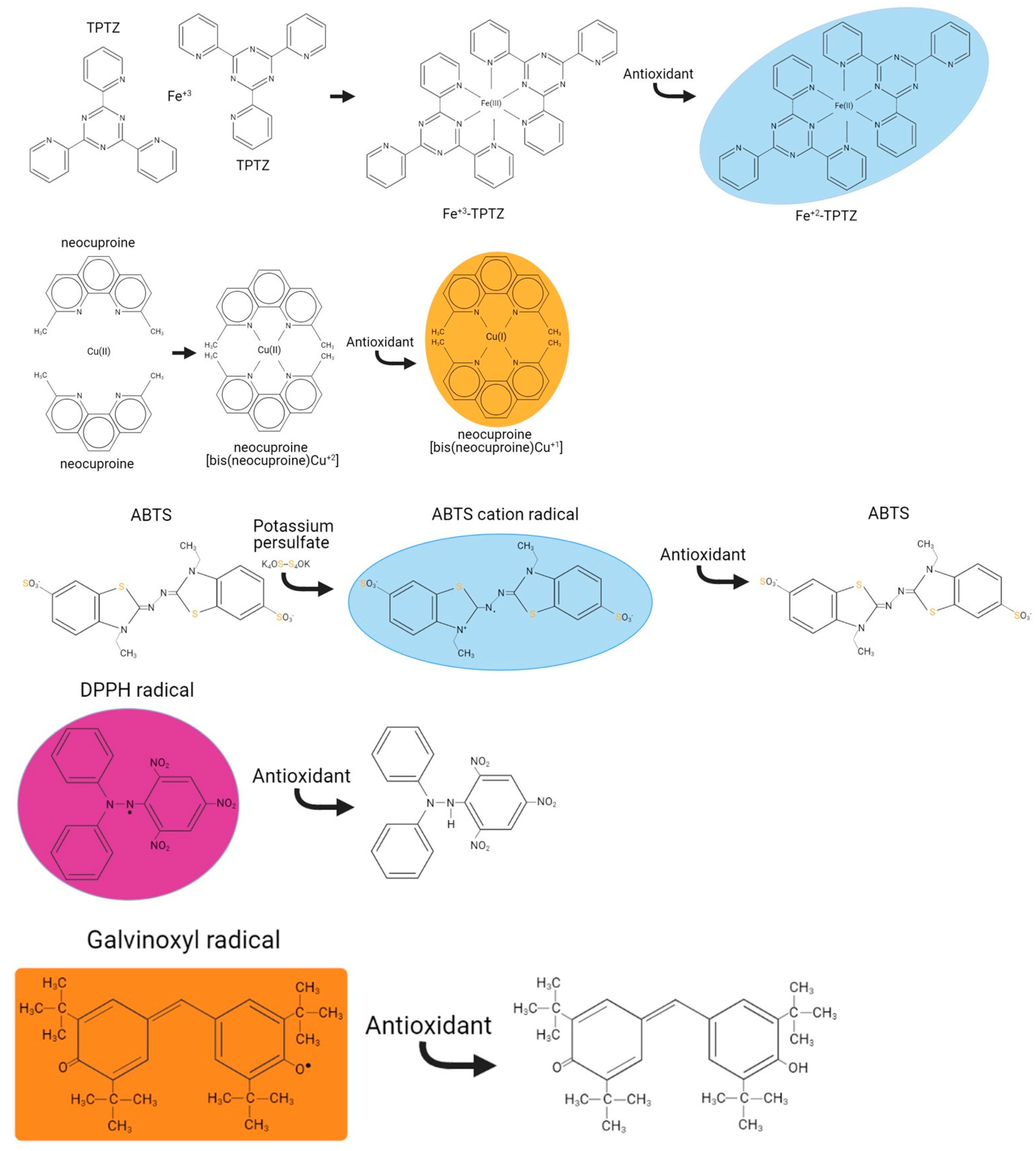
Figure 1) was based on assays using the 2,4,6-tri-pyridyl-s-triazine [
16] and neocuproine reagents [
17], respectively. Both assays require first the metal cation to generate a complex with the reagent, followed by the reduction of the metal cation to generate a chromogenic reduced metal-reagent complex. Finally, assays based on the reduction of the stable DPPH [
18], ABTS
•+ cation [
19] and galvinoxyl radicals are performed using the loss of their colour when scavenged by a phytochemical extract, to assess the % of scavenging capacity (
Figure 1). Overall, the protocols presented underwent extensive troubleshooting and optimisation of the conditions existing in the original methods, previously applied on extracts from the sumac plant [
20] aiming to provide a unified gold standard approach.
4. Expected Results
The multiparametric protocol described here can be applied to any type of plant material. Previously, we demonstrated this approach in our article on the sumac root, leaf and stem extracts on an in vitro ethanol toxicity model system [
20]. In this study, we provide the biochemical analysis of Greek herbal plant extracts (dataset 1) and other common plants (dataset 2) summarised as indicative exemplars of our outlined approach in
Supplementary Excel files with automated calculations to assist the end user. Each sheet contains a respective protocol in addition to a standard curve and sample analysis (for four replicates per samples). The technical reproducibility of the methods presented is reflected on the low coefficient of variance obtained from independent replicates of each sample. This is a result of the simplicity of each assay performed in a minimum number of steps. Simple statistics with unpaired tests can be performed using the Excel files provided. Furthermore, the results can be collectively processed (in their averages per sample) after z-score standardisation to avoid the large differences among data skewing the findings. The results are easily presented in radar charts (
Figure 3), which can collectively summarise the parameters measured for a great number of plants. Values were z-score standardised as outlined in the automated Excel file. In the first dataset of the Greek herbal plants, the stinging nettle (
Urtica urens, Uu) is the sample in the inner center of the radar charts, thus with the minimum amount of all measurement parameters, while for other samples characteristic increased values such as tannins for
Laurus nobilis (Ln) can be easily visualised.
Although there are many biochemical methods available for determining the antioxidant activities of biological materials [
27], to our knowledge there has been no attempt so far in the research community to generate a gold standard approach on the outlined topic of a general characterisation of plant extracts. Furthermore, as a general rule for the assays available in the literature, these mostly rely on large reagent and sample volumes, which in turn results in increased consumption of reagents, generating additional lab waste and requiring more samples, which, in turn, would reflect on the sensitivity of the methods [
28]. The methods developed here aim to quickly, accurately, and with the highest sensitivity provide results for the general characterisation of plants.
The protocols presented here provide a detailed characterisation of plant extract as their primary focus to identify the main phytochemical categories. However, the specific identification of compounds would require significant and cumbersome analytical instrumentation such as chromatographic separation coupled with mass spectrometry or NMR spectroscopy to achieve the specific elucidation of plant extract chemical composition [
29]. Although there have been many advances in hyphenated analytical methods for the quantification and identification of polyphenols, flavonoids and tannins, providing their analytical coverage is limited by matrix effects and the cost of analytical equipment [
30]. The protocol presented here could also be part of a more detailed assessment if a user chooses to reconstitute the samples from step 4 following their extraction and proceed towards an HPLC-MS analysis.
All methods presented here have been modified extensively from their original studies and were further modulated and optimised to promote an analytical approach with the least number of steps and for processing large sample numbers, with results comparable in reproducibility, accuracy and applicability to a number of kits reviewed. The protocols presented result in significant time saving, reducing the total cost for reagents and providing a cost-effective method. As an example, the typical kits available would be limited to usually 200 samples and cost approximately 500 euros for FeRP and polyphenols. The flexibility of the multiparametric protocol also highlights its versatility as different parameters can be handled and the workload can be spread appropriately, or users can focus only on specific methods of their research interest. The feasibility of this protocol is reflected in the
Supplementary Excel files, which provide worked-out examples for the fast processing of results generated. All calculations and analyses are easily performed in an automated fashion which allows the user to generate their quantitative results. We foresee the application of this method to the wider research community and a diverse set of research themes such as phytochemistry, toxicology and redox biochemistry.
6. Reagents Setup
Reagents are listed in order of appearance for each protocol.
Amount of 2% NaNO2: Prepare fresh by dissolving 200 mg NaNO2 in 10 mL ddH2O.
Amount of 7.5% AlCl3: Prepare fresh by dissolving 750 mg AlCl3 in 10 mL ddH2O. !CAUTION Dissolution is exothermal and produces gas and all tasks must be performed in the fume hood.
Amount of 3.5 M NaOH: Dissolve 14 g NaOH (MW: 40 g/mol) in 100 mL ddH2O.
Amount of 4% vanillin: Prepare fresh by dissolving 400 mg vanillin in 10 mL methanol.
Amount of 100% H2SO4: CAUTION! H2SO4 is concentrated acid and all handling is performed in the fume hood.
Amount of 4x Folin reagent: Prepare fresh by diluting the Folin reagent 4x with ddH2O.
Amount of 1.89 M Na2CO3: Dissolve 10 g Na2CO3 (MW: 105.99 g/mol) in 50 mL ddH2O.
Amount of 300 mM acetic acid: Prepare fresh by dissolving 0.093 g sodium acetate anhydrous (MW: 82.03 g/mol) and 0.8 mL glacial acetic acid in 49.2 mL ddH2O.
Amount of 40 mM HCl: Diluting the concentrated 37% (or 12 M) HCl 300x with ddH2O by mixing 150 mL ddH2O with 0.5 mL 12 M HCl under stirring. CAUTION! HCl is a concentrated fumigous acid and all handling must be performed in the fume hood.
TPTZ: Prepare fresh by dissolving 3.12 mg TPTZ (MW: 312.33 g/mol) in 500 μL methanol. Then add 500 μL 40 mM HCl.
Amount of 0.54% FeCl3.6H2O: Prepare fresh by dissolving 54 mg ferric chloride hexahydrate in 10 mL ddH2O.
FeRP reagent: Mix TPTZ:0.54% FeCl
3.6H
2O:300 mM acetic acid in a ratio of 1:1:10. This complex form of iron when reduced will absorb at 595 nm (
Figure 1).
Amount of 10 mM Cu+2: Prepare fresh by dissolving 24.9 mg copper sulphate pentahydrate (MW: 249.68 g/mL) in 10 mL ddH2O.
Amount of 6 mM neocuproine: Prepare fresh by dissolving 16 mg neocuproine (MW: 262.73 g/mL) in 10 mL ddH2O.
1M ammonium acetate: Prepare fresh by dissolving 3.86 g ammonium acetate (MW: 77.08 g/mol) in 25 mL ddH2O and after complete dissolution, adjust volume to 50 mL. !CAUTION The solid ammonium acetate will occupy some volume of the solution.
Cu-neocuproine-ammonium acetate reagent: Mix 10 mM Cu
+2, 6 mM neocuproine and 1 M ammonium acetate in equal volumes (1:1:1). This complex form of copper when reduced will absorb at 450 nm (
Figure 1).
Amount of 100 mM acetic acid pH 5.5: Prepare fresh by dissolving 0.42 g sodium acetate anhydrous (MW: 82.04 g/mol) in ddH2O. Adjust the pH to 5.5 and the final volume to 50 mL.
Methanol:100 mM acetic acid pH 5.5 (9:1): Prepare fresh by mixing 80 mL methanol with 20 mL 100 mM acetic acid pH 5.5.
DPPH radical: Prepare fresh by dissolving 25 mg DPPH (MW: 394.32 g/mol) in 10 mL methanol. Dilute this stock with methanol:acetic acid pH 5.5 (8:2) and filter (0.22 μM). Dilute further this stock with methanol:acetic acid pH 5.5 (8:2) to a value of ~1.3A at 515 nm. This value is set to be in the linear range of the spectrophotometer/microplate when diluted 1.5× in the assay (mixing 200 μL from the DPPH with 100 μL ddH
2O). DPPH is a stable radical which absorbs at 515 nm and when scavenged decolourises (
Figure 1).
Amount of 14 mM ABTS radical cation (ABTS•+): Prepare fresh by dissolving 77 mg ABTS (MW: 548.68 g/mol) in 10 mL ddH2O.
Amount of 5 mM potassium persulphate: Prepare fresh by dissolving 13.5 mg potassium persulphate (MW: 270.32 g/mol) in 10 mL ddH2O.
ABTS radical cation (ABTS•+): Mix the 14 mM ABTS and the 5 mM potassium persulphate reagents 1:1 and incubate in the dark at RT for 12 h before use. ABTS
•+ is a stable radical formed by potassium persulphate radical initiation. Dilute further this stock with ddH
2O to a value of ~1.3 A at 734 nm. This value is set to be in the linear range of the spectrophotometer/microplate when diluted 1.5× in the assay (mixing 200 μL from the ABTS radical cation with 100 μL ddH
2O). ABTS
•+ is a stable radical formed by potassium persulphate radical initiation which absorbs at 734 nm and when scavenged decolourises (
Figure 1).
Amount of 100 mM citric acid pH 6: Prepare fresh by dissolving 0.1921 g sodium acetate anhydrous (MW: 192.12 g/mol) in ddH2O. Adjust the pH to 6 and the final volume to 100 mL.
Methanol:100 mM citric acid pH 6 (9:1): Prepare fresh by mixing 90 mL methanol with 10 mL 100 mM citric acid pH 6.
Galvinoxyl radical: Prepare fresh by dissolving 50 mg DPPH (MW: 394.32 g/mol) in 10 mL methanol. Dilute this stock with methanol:citric acid pH 6 (9:1) and filter (0.22 μM). Dilute further this stock with methanol:citric acid pH 6 (9:1) to a value of ~1.3 A at 435 nm. This value is set to be in the linear range of the spectrophotometer/microplate when diluted 1.5x in the assay [mixing 200 μL from the galvinoxyl radical with 100 μL methanol:citric acid pH 6 (9:1)]. Galvinoxyl radical is a stable radical which absorbs at 435 nm and when scavenged decolourises (
Figure 1).
Standard Curves for Assays
Catechin: Prepare a 30 mM stock solution in methanol by dissolving 8.79 mg catechin (MW: 290.27 g/mol) in 1 mL methanol. Dilute the 30 mM stock 300x to 100 μM and follow to 10 μM in ddH2O. Prepare a series of dilutions of standards from each stock for 1–10 μM and 10–100 μΜ, respectively, for the linear standard curves.
Gallic acid: Prepare a 10 mM stock solution by dissolving 17.1 mg gallic acid (MW: 170.12 g/mol) in 10 mL methanol. Dilute the 10 mM stock solution to 100 μM and follow to 10 μM in ddH2O. Prepare a series of dilutions of standards from each stock for 1–10 μM and 10–100 μΜ, respectively, for the linear standard curves.
















{kind=link}
{kind=link}
{kind=link}