
An Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry Method with Online Solid-Phase Extraction Sample Preparation for the High-Throughput and Sensitive Determination of Ostarine in Human Urine



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents and Solutions
2.2. Urine Samples
2.3. Instrumentation
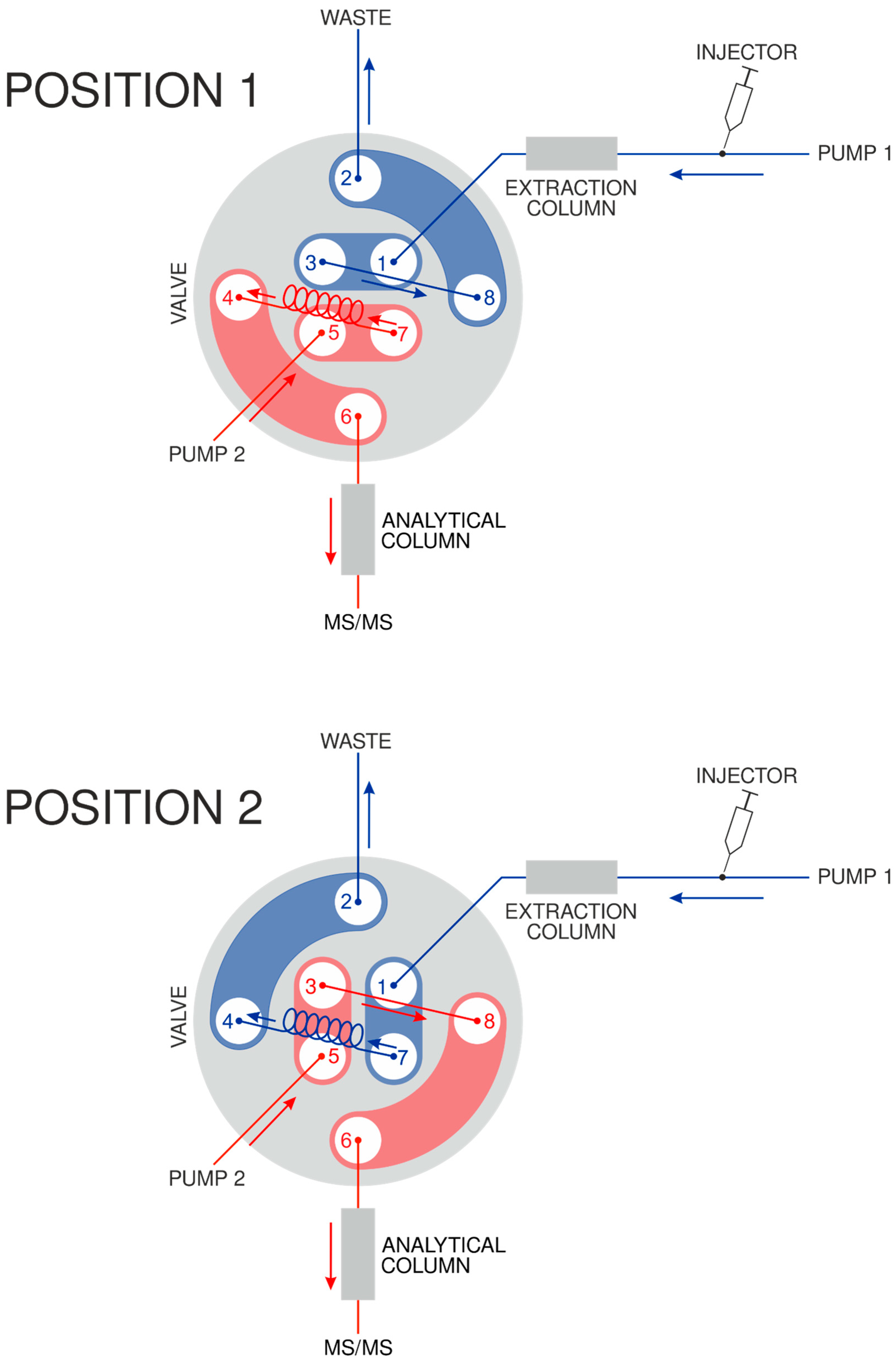
2.4. Online Solid-Phase Extraction (Online-SPE)
2.5. Method Validation
2.5.1. Linearity and Range
2.5.2. Limit of Quantification (LOQ)
2.5.3. Limit of Detection (LOD)
2.5.4. Accuracy and Precision
2.5.5. Recovery and Matrix Effects
2.5.6. Carryover
2.5.7. Selectivity and Robustness
2.6. WADA Criteria for Method Application
2.6.1. Chromatographic Criteria
2.6.2. Mass Spectrometric Criteria
3. Results and Discussion
3.1. Method Optimization
3.1.1. MS/MS Parameters
3.1.2. Online-SPE and UHPLC Parameters
3.2. Method Validation
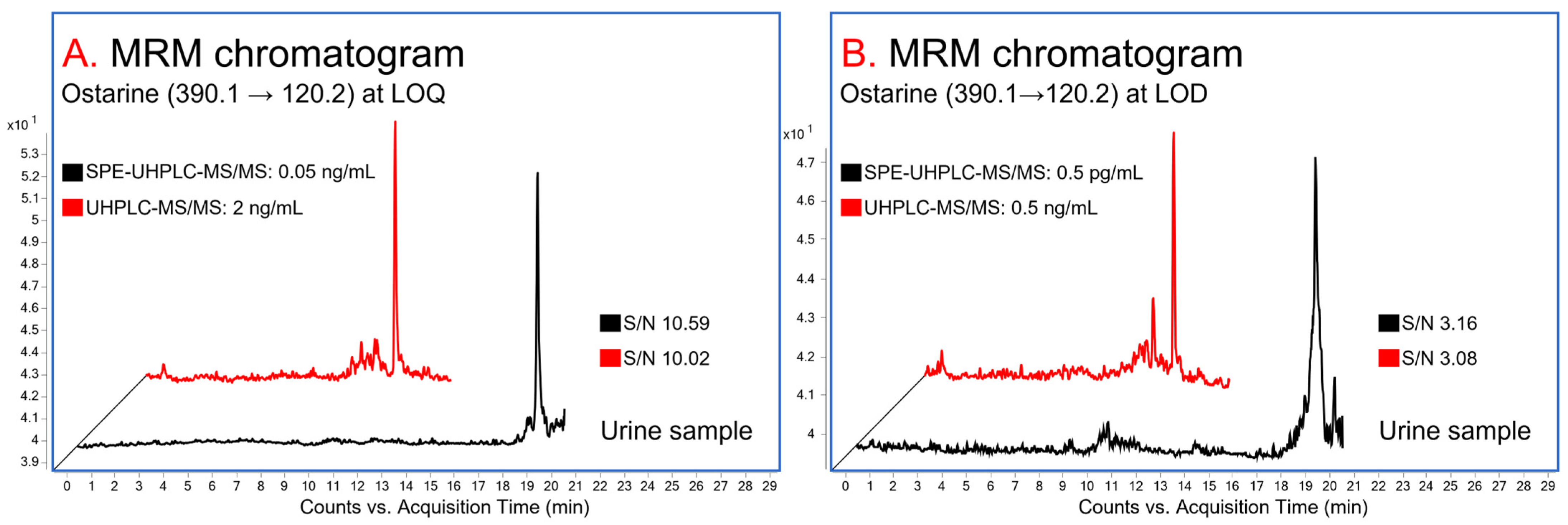
3.2.1. Linearity, Range, and LOQ
3.2.2. LOD
3.2.3. Accuracy and Precision
3.2.4. Recovery and Matrix Effect
3.2.5. Carryover
3.2.6. Selectivity and Robustness
3.2.7. Comparison of the Validation Parameters: SPE-UHPLC-MS/MS vs. UHPLC-MS/MS
3.3. Method Application and Fulfilment of WADA Criteria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Gillece, J.D.; Folkerts, M.; Reinning, L.; et al. A Phase II Clinical Trial of Pembrolizumab and Enobosarm in Patients with Androgen Receptor-Positive Metastatic Triple-Negative Breast Cancer. Oncologist 2021, 26, 99-e217. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, S.; Krishnan, V.; Storer, T.W.; Steiner, M.; Dobs, A.S. Androgens and Selective Androgen Receptor Modulators to Treat Functional Limitations Associated With Aging and Chronic Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2023, 78, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Dobs, A.S.; Boccia, R.V.; Croot, C.C.; Gabrail, N.Y.; Dalton, J.T.; Hancock, M.L.; Johnston, M.A.; Steiner, M.S. Effects of enobosarm on muscle wasting and physical function in patients with cancer: A double-blind, randomised controlled phase 2 trial. Lancet Oncol. 2013, 14, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Dalton, J.T.; Barnette, K.G.; Bohl, C.E.; Hancock, M.L.; Rodriguez, D.; Dodson, S.T.; Morton, R.A.; Steiner, M.S. The selective androgen receptor modulator GTx-024 (enobosarm) improves lean body mass and physical function in healthy elderly men and postmenopausal women: Results of a double-blind, placebo-controlled phase II trial. J. Cachexia Sarcopenia Muscle 2011, 2, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, E.; Ajazi, E.; Wang, X.; Johnston, M.A.M.; Crawford, J. Predictors of Physical and Functional Loss in Advanced-Stage Lung Cancer Patients Receiving Platinum Chemotherapy. J. Thorac. Oncol. 2018, 13, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Efimenko, I.V.; Valancy, D.; Dubin, J.M.; Ramasamy, R. Adverse effects and potential benefits among selective androgen receptor modulators users: A cross-sectional survey. Int. J. Impot. Res. 2022, 34, 757–761. [Google Scholar] [CrossRef]
- Bedi, H.; Hammond, C.; Sanders, D.; Yang, H.M.; Yoshida, E.M. Drug-Induced Liver Injury From Enobosarm (Ostarine), a Selective Androgen Receptor Modulator. ACG Case Rep. J. 2021, 8, e00518. [Google Scholar] [CrossRef]
- Kintz, P.; Gheddar, L.; Paradis, C.; Chinellato, M.; Ameline, A.; Raul, J.S.; Oliva-Labadie, M. Peroxisome Proliferator-Activated Receptor Delta Agonist (PPAR-δ) and Selective Androgen Receptor Modulator (SARM) Abuse: Clinical, Analytical and Biological Data in a Case Involving a Poisonous Combination of GW1516 (Cardarine) and MK2866 (Ostarine). Toxics 2021, 9, 251. [Google Scholar] [CrossRef]
- Koller, T.; Vrbova, P.; Meciarova, I.; Molcan, P.; Smitka, M.; Adamcova Selcanova, S.; Skladany, L. Liver injury associated with the use of selective androgen receptor modulators and post-cycle therapy: Two case reports and literature review. World J. Clin. Cases 2021, 9, 4062–4071. [Google Scholar] [CrossRef]
- Weinblatt, D.; Roy, S. Drug-Induced Liver Injury Secondary to Enobosarm: A Selective Androgen Receptor Modulator. J. Med. Cases 2022, 13, 244–248. [Google Scholar] [CrossRef]
- Leciejewska, N.; Pruszyńska-Oszmałek, E.; Nogowski, L.; Sassek, M.; Strowski, M.Z.; Kołodziejski, P.A. Sex-specific cytotoxicity of ostarine in cardiomyocytes. Mol. Cell Endocrinol. 2023, 577, 112037. [Google Scholar] [CrossRef]
- Mertens, J.E.; Bömmer, M.T.C.; Regier, M.B.; Gabriëls, G.; Pavenstädt, H.; Grünewald, I.; Horvath, J.; Trebicka, J.; Schmidt, H.; Schlevogt, B. Liver Injury after Selective Androgen Receptor Modulator Intake: A Case Report and Review of the Literature. Z. Gastroenterol. 2023. ahead of print. [Google Scholar] [CrossRef]
- World Anti-Doping Agency (WADA). World Anti-Doping Code 2021. Article 4.3.1.3. Available online: https://www.wada-ama.org/sites/default/files/resources/files/2021_wada_code.pdf (accessed on 9 November 2023).
- Machek, S.B.; Cardaci, T.D.; Wilburn, D.T.; Willoughby, D.S. Considerations, possible contraindications, and potential mechanisms for deleterious effect in recreational and athletic use of selective androgen receptor modulators (SARMs) in lieu of anabolic androgenic steroids: A narrative review. Steroids 2020, 164, 108753. [Google Scholar] [CrossRef]
- World Anti-Doping Agency (WADA). 2019 Anti-Doping Testing Figures. Available online: https://www.wada-ama.org/sites/default/files/resources/files/2019_anti-doping_testing_figures_en.pdf (accessed on 9 November 2023).
- Görgens, C.; Guddat, S.; Thomas, A.; Wachsmuth, P.; Orlovius, A.K.; Sigmund, G.; Thevis, M.; Schänzer, W. Simplifying and expanding analytical capabilities for various classes of doping agents by means of direct urine injection high performance liquid chromatography high resolution/high accuracy mass spectrometry. J. Pharm. Biomed. Anal. 2016, 131, 482–496. [Google Scholar] [CrossRef]
- Thevis, M.; Thomas, A.; Möller, I.; Geyer, H.; Dalton, J.T.; Schänzer, W. Mass spectrometric characterization of urinary metabolites of the selective androgen receptor modulator S-22 to identify potential targets for routine doping controls. Rapid Commun. Mass Spectrom. 2011, 25, 2187–2195. [Google Scholar] [CrossRef]
- Sobolevsky, T.; Ahrens, B. High-throughput liquid chromatography tandem mass spectrometry assay as initial testing procedure for analysis of total urinary fraction. Drug Test. Anal. 2021, 13, 283–298. [Google Scholar] [CrossRef]
- Gadaj, A.; Ventura, E.; Healy, J.; Botrè, F.; Sterk, S.S.; Buckley, T.; Mooney, M.H. Enhanced UHPLC-MS/MS screening of selective androgen receptor modulators following urine hydrolysis. MethodsX 2020, 7, 100926. [Google Scholar] [CrossRef]
- Walpurgis, K.; Rubio, A.; Wagener, F.; Krug, O.; Knoop, A.; Görgens, C.; Guddat, S.; Thevis, M. Elimination profiles of microdosed ostarine mimicking contaminated products ingestion. Drug Test. Anal. 2020, 12, 1570–1580. [Google Scholar] [CrossRef]
- Stacchini, C.; Botrè, F.; Comunità, F.; de la Torre, X.; Dima, A.P.; Ricci, M.; Mazzarino, M. Simultaneous detection of different chemical classes of selective androgen receptor modulators in urine by liquid chromatography-mass spectrometry-based techniques. J. Pharm. Biomed. Anal. 2021, 195, 113849. [Google Scholar] [CrossRef]
- Temerdashev, A.; Dmitrieva, E.; Azaryan, A.; Gashimova, E. A novel approach to the quantification of urinary aryl-propionamide-derived SARMs by UHPLC-MS/MS. Biomed. Chromatogr. 2020, 34, e4700. [Google Scholar] [CrossRef]
- Guddat, S.; Solymos, E.; Orlovius, A.; Thomas, A.; Sigmund, G.; Geyer, H.; Thevis, M.; Schänzer, W. High-throughput screening for various classes of doping agents using a new ‘dilute-and-shoot’ liquid chromatography-tandem mass spectrometry multi-target approach. Drug Test. Anal. 2011, 3, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Viette, V.; Fathi, M.; Rudaz, S.; Hochstrasser, D.; Veuthey, J.L. Current role of liquid chromatography coupled to mass spectrometry in clinical toxicology screening methods. Clin. Chem. Lab. Med. 2011, 49, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Schänzer, W. Mass spectrometry of selective androgen receptor modulators. J. Mass Spectrom. 2008, 43, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Gerace, E.; Thomas, A.; Beuck, S.; Geyer, H.; Schlörer, N.; Kearbey, J.D.; Dalton, J.T.; Schänzer, W. Characterization of in vitro generated metabolites of the selective androgen receptor modulators S-22 and S-23 and in vivo comparison to post-administration canine urine specimens. Drug Test. Anal. 2010, 2, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Thevis, M.; Thomas, A.; Fusshöller, G.; Beuck, S.; Geyer, H.; Schänzer, W. Mass spectrometric characterization of urinary metabolites of the selective androgen receptor modulator andarine (S-4) for routine doping control purposes. Rapid Commun. Mass Spectrom. 2010, 24, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Grata, E.; Perrenoud, L.; Saugy, M.; Baume, N. SARM-S4 and metabolites detection in sports drug testing: A case report. Forensic Sci. Int. 2011, 213, 104–108. [Google Scholar] [CrossRef]
- Starcevic, B.; Ahrens, B.D.; Butch, A.W. Detection of the selective androgen receptor modulator S-4 (Andarine) in a doping control sample. Drug Test. Anal. 2013, 5, 377–379. [Google Scholar] [CrossRef]
- Airado-Rodríguez, D.; Cruces-Blanco, C.; García-Campaña, A.M. Dispersive liquid-liquid microextraction prior to field-amplified sample injection for the sensitive analysis of 3,4-methylenedioxymethamphetamine, phencyclidine and lysergic acid diethylamide by capillary electrophoresis in human urine. J. Chromatogr. A 2012, 1267, 189–197. [Google Scholar] [CrossRef]
- World Anti-Doping Agency (WADA). International Standard for Laboratories 2021. Available online: https://www.wada-ama.org/sites/default/files/resources/files/isl_2021.pdf (accessed on 1 September 2023).
- World Anti-Doping Agency (WADA). TD2021IDCR. Available online: https://www.wada-ama.org/sites/default/files/resources/files/td2021idcr_final_eng_0.pdf (accessed on 1 September 2023).
- World Anti-Doping Agency (WADA). TD2022MRPL. Available online: https://www.wada-ama.org/sites/default/files/2022-01/td2022mrpl_v1.1_eng_0.pdf (accessed on 1 September 2023).



{kind=link}
{kind=link}
| Time (min) | A * (%) | B ** (%) | Flow (mL/min) | Process | |
|---|---|---|---|---|---|
| Binary pump 1 | |||||
| 1 | 1.00 | 95 | 5 | 0.5 | Loading |
| 2 | 1.01 | 35 | 65 | 1.5 | Extraction |
| 3 | 3.50 | 35 | 65 | 1.5 | |
| 4 | 3.51 | 35 | 65 | 0.5 | Position 2: 4.60–5.45 min |
| 5 | 5.45 | 35 | 65 | 0.5 | |
| 6 | 5.46 | 95 | 5 | 0.5 | Re-equilibrating |
| 7 | 20.00 | 95 | 5 | 0.5 | |
| Binary pump 2 | |||||
| 1 | 10.30 | 95 | 5 | 0.5 | Separating |
| 2 | 12.30 | 5 | 95 | 0.5 | |
| 3 | 20.00 | 5 | 95 | 0.5 | |
| Loading | Washing | Peak Area ** (%) MRM (m/z): 390.1 → 120.2 | ||
|---|---|---|---|---|
| Flow (mL/min) | Elution | B * (%) | Flow (mL/min) | |
| 0.5 | Gradient | 5–65% in 2.50 min | 1.5 | 33.3 |
| 0.5 | Isocratic | 65% for 2.50 min | 1.5 | 100 |
| 0.5 | Isocratic | 65% for 2.50 min | 0.5 | 65.7 |
| 1.5 | Isocratic | 65% for 2.50 min | 1.5 | 85.2 |
| Mobile Phase Composition | Peak Area * (%) MRM (m/z): 390.1 → 120.2 | |
|---|---|---|
| Online-SPE | UHPLC | |
| 0.1% FA | 0.1% FA | 100 |
| 0.1% FA | 1 mM AF + 0.1% FA | 90.3 |
| 1 mM AF + 0.1% FA | 0.1% FA | 95.6 |
| 1 mM AF + 0.1% FA | 1 mM AF + 0.1% FA | 85.9 |
| 0.1% AA | 0.1% AA | 78.1 |
| 0.1% AA | 1 mM Ac + 0.1% AA | 83.4 |
| 1 mM Ac + 0.1% AA | 0.1% AA | 80.9 |
| 1 mM Ac + 0.1% AA | 1 mM Ac + 0.1% AA | 82.1 |
| Parameter | SPE-UHPLC-MS/MS * | UHPLC-MS/MS ** |
|---|---|---|
| Linear range (ng/mL) | 0.05 to 25 | 2 to 1000 |
| Slope (a) | 0.3925 | 0.0276 |
| SDa (n = 18) | 0.0019 | 0.0001 |
| Intercept (b) | −0.0360 | −0.0427 |
| SDb (n = 18) | 0.0194 | 0.0414 |
| r2 | 0.9999 | 0.9999 |
| LOQ *** (ng/mL) | 0.05 | 2 |
| LOD **** (ng/mL) | 0.0005 | 0.5 |
| QC Samples | Intra-Day (n = 5) | Inter-Day (n = 15) | ||||
|---|---|---|---|---|---|---|
| cN (ng/mL) | cF (ng/mL) | RE (%) | RSD (%) | cF (ng/mL) | RE (%) | RSD (%) |
| SPE-UHPLC-MS/MS * | ||||||
| 0.25 | 0.27 | +7.5 | 1.9 | 0.26 | +2.8 | 4.5 |
| 1.25 | 1.29 | +3.5 | 0.8 | 1.27 | +1.6 | 4.2 |
| 12.50 | 12.77 | +2.1 | 0.9 | 12.91 | +3.3 | 1.7 |
| UHPLC-MS/MS ** | ||||||
| 5 | 5.79 | +15.9 | 3.9 | 5.59 | +11.7 | 4.7 |
| 25 | 26.35 | +5.4 | 1.7 | 26.34 | +5.4 | 3.9 |
| 250 | 276.50 | +10.6 | 1.7 | 281.96 | +12.8 | 2.9 |
| Urine/Reference Samples (n = 5) | |||
|---|---|---|---|
| c (ng/mL) | Recovery (%) | ME without IS (%) | ME with IS (%) |
| SPE-UHPLC-MS/MS * | |||
| 0.25 | 102.3 | −15.0 | +3.3 |
| 1.25 | 98.7 | −28.5 | −1.4 |
| 12.50 | 96.0 | −40.0 | −4.0 |
| UHPLC-MS/MS ** | |||
| 5 | 81.3 | −23.5 | −23.7 |
| 25 | 76.5 | −32.8 | −24.7 |
| 250 | 75.3 | −34.4 | −24.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slíž, K.; Piešťanský, J.; Mikuš, P. An Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry Method with Online Solid-Phase Extraction Sample Preparation for the High-Throughput and Sensitive Determination of Ostarine in Human Urine. Methods Protoc. 2024, 7, 10. https://doi.org/10.3390/mps7010010
Slíž K, Piešťanský J, Mikuš P. An Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry Method with Online Solid-Phase Extraction Sample Preparation for the High-Throughput and Sensitive Determination of Ostarine in Human Urine. Methods and Protocols. 2024; 7(1):10. https://doi.org/10.3390/mps7010010
Chicago/Turabian StyleSlíž, Kristián, Juraj Piešťanský, and Peter Mikuš. 2024. "An Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry Method with Online Solid-Phase Extraction Sample Preparation for the High-Throughput and Sensitive Determination of Ostarine in Human Urine" Methods and Protocols 7, no. 1: 10. https://doi.org/10.3390/mps7010010
APA StyleSlíž, K., Piešťanský, J., & Mikuš, P. (2024). An Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry Method with Online Solid-Phase Extraction Sample Preparation for the High-Throughput and Sensitive Determination of Ostarine in Human Urine. Methods and Protocols, 7(1), 10. https://doi.org/10.3390/mps7010010