
Transcriptomic Analysis and Functional Gene Expression in Different Stages of Gonadal Development of Macrobrachium rosenbergii

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Total RNA Extraction and Transcriptome Sequencing
2.3. Sequence Assembly and Gene Function Annotation
2.4. Identification of Differentially Expressed Genes, and GO and KEGG Enrichment Analysis
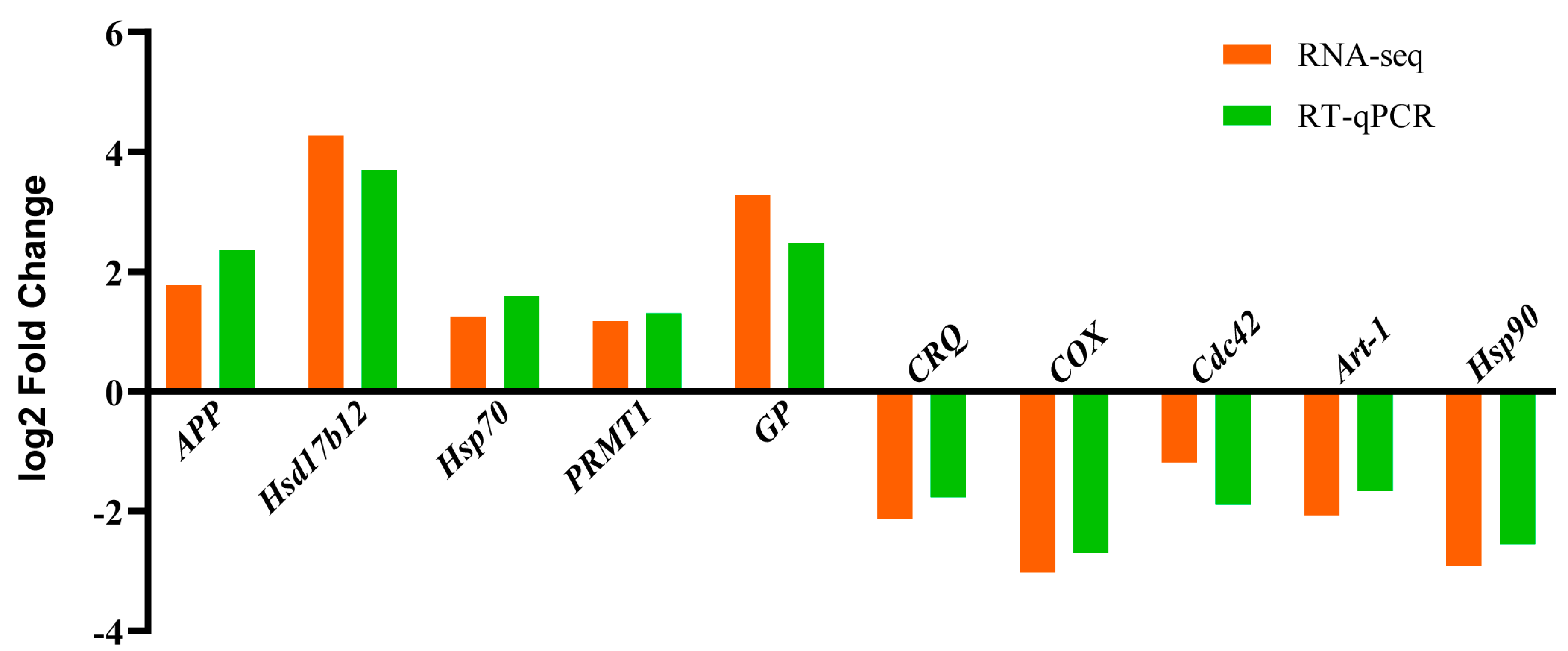
2.5. Real-Time Quantitative PCR Validation of Differentially Expressed Genes
3. Results
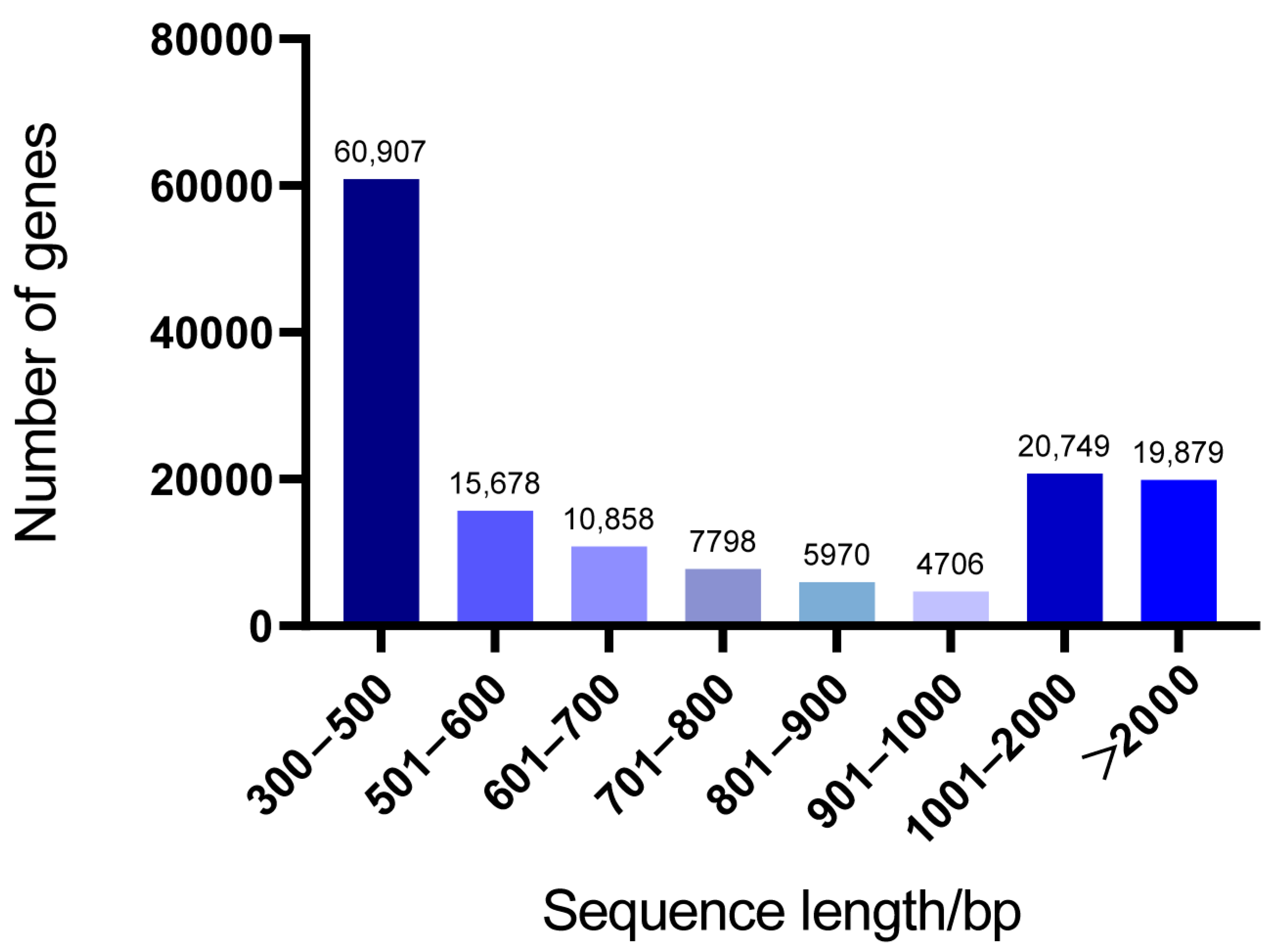
3.1. Transcript Assembly and Statistical Analysis
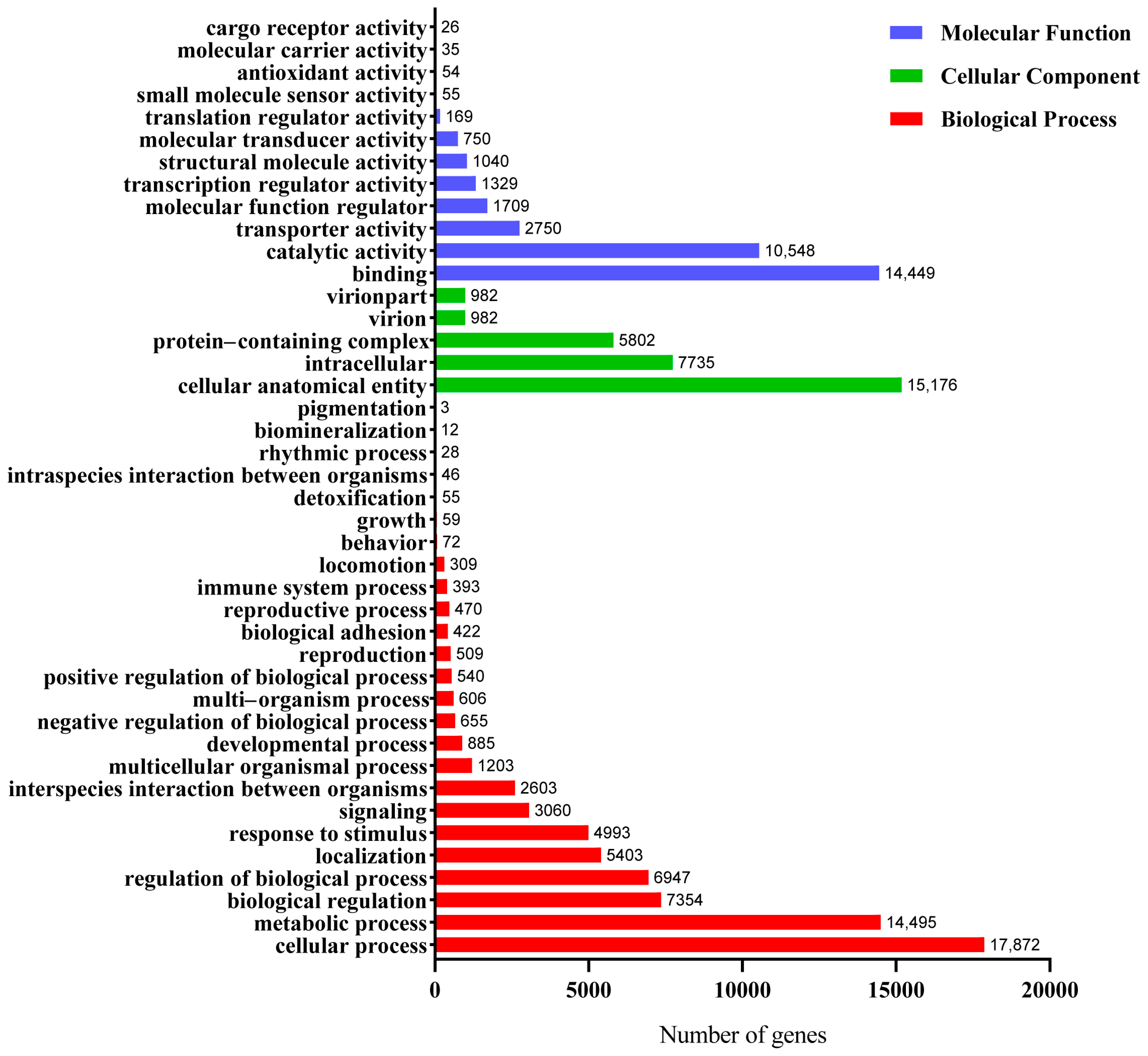
3.2. Functional Annotation and Classification of the Unigenes
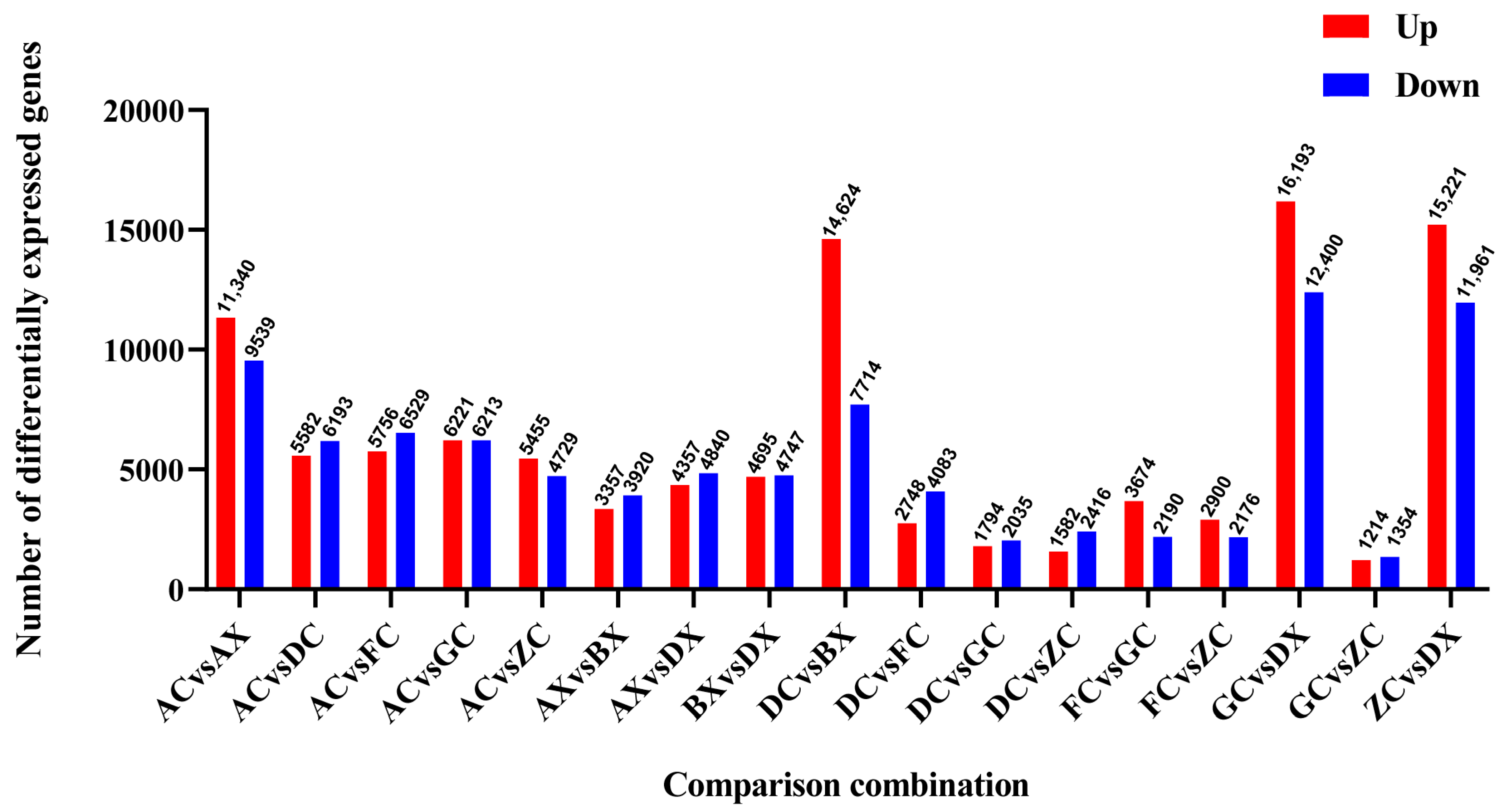
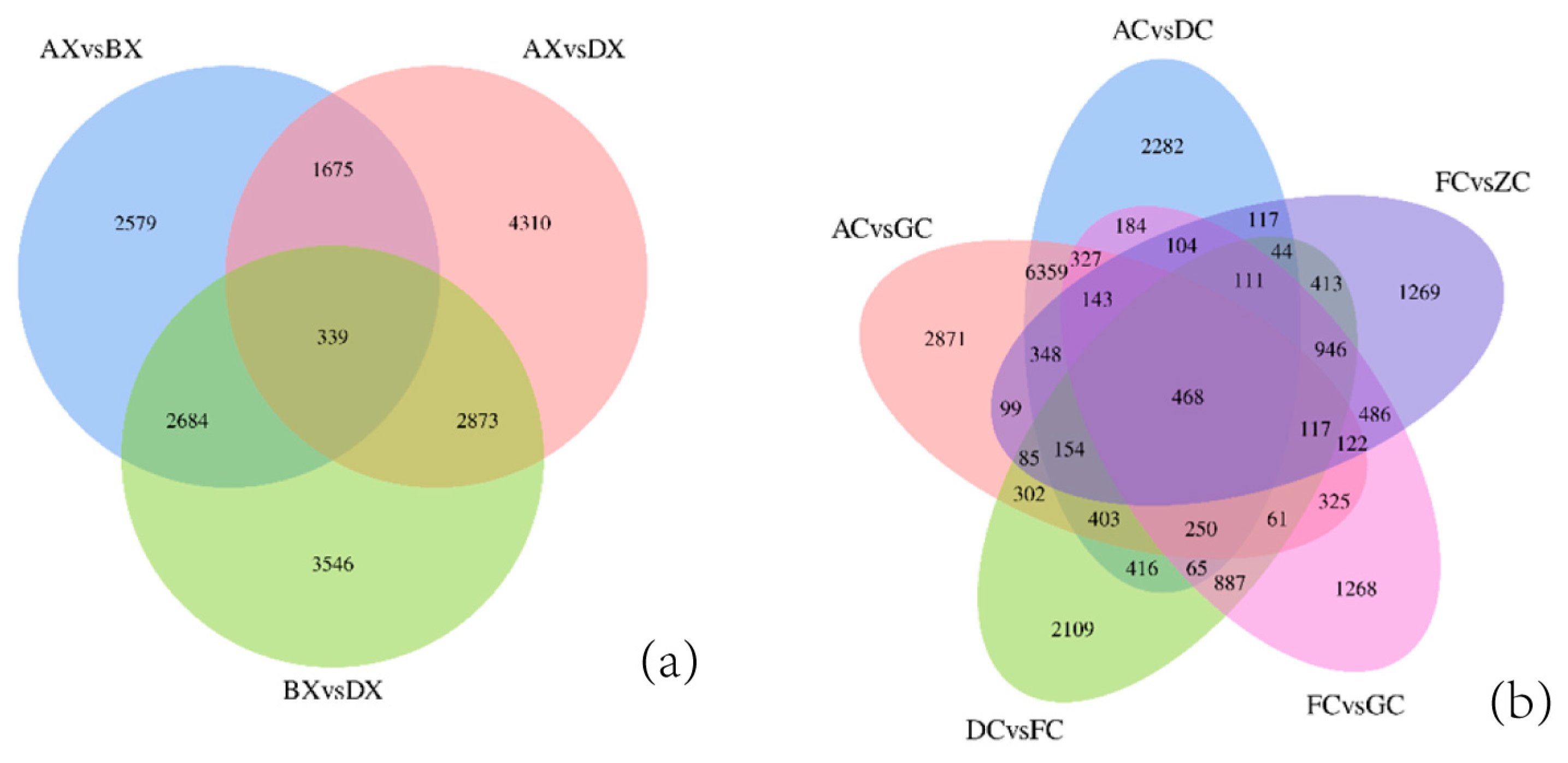
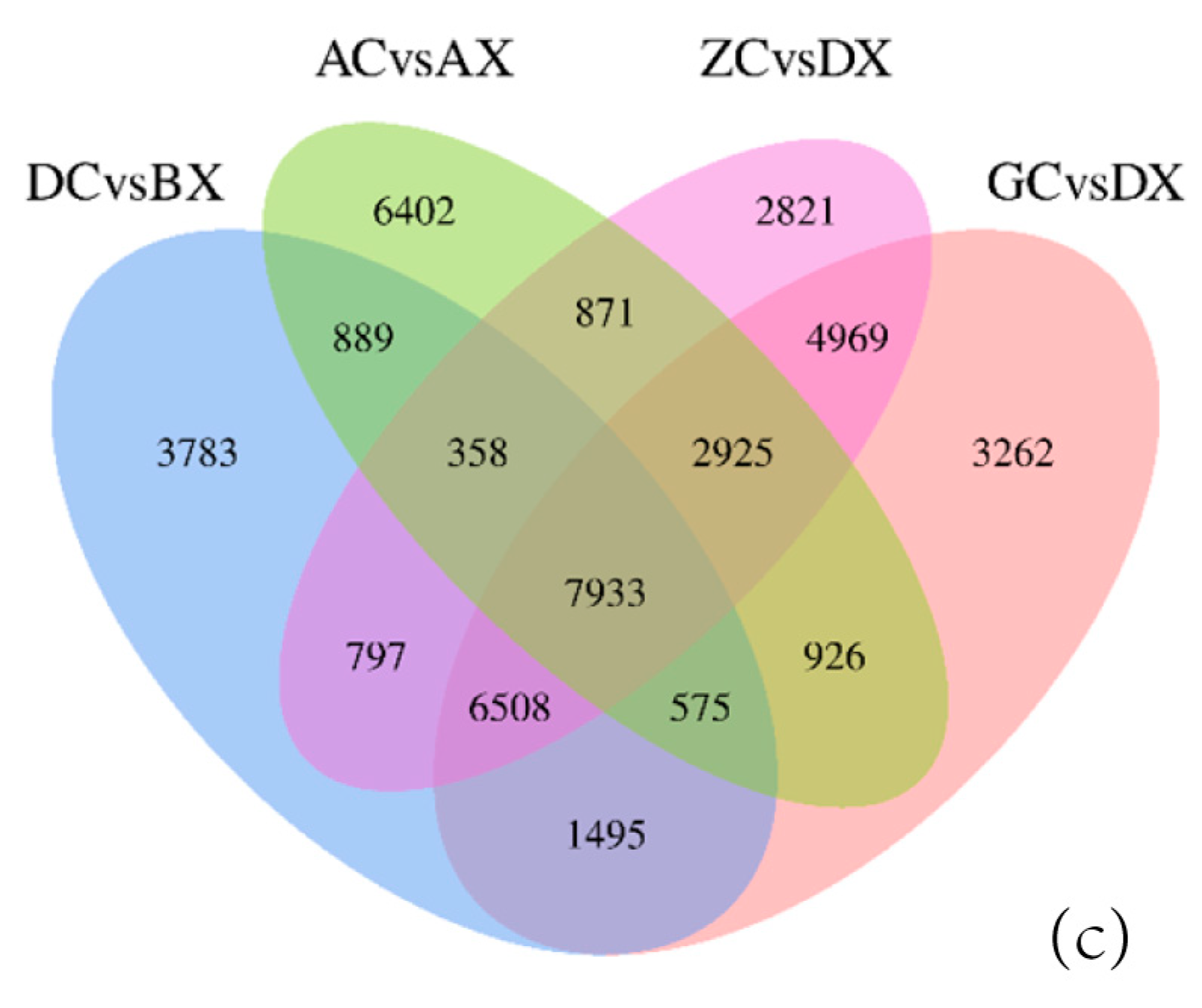
3.3. Identification of Differentially Expressed Genes
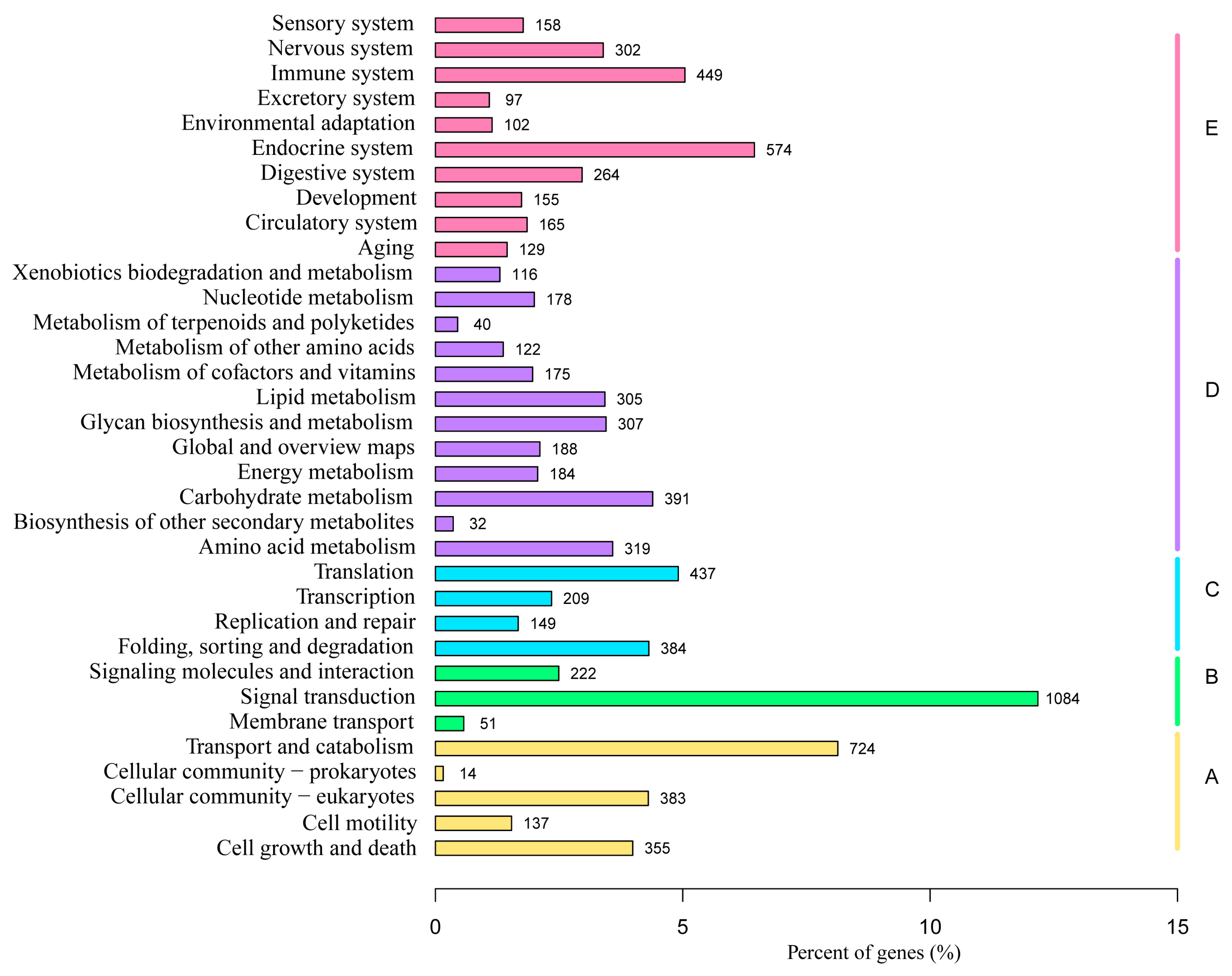
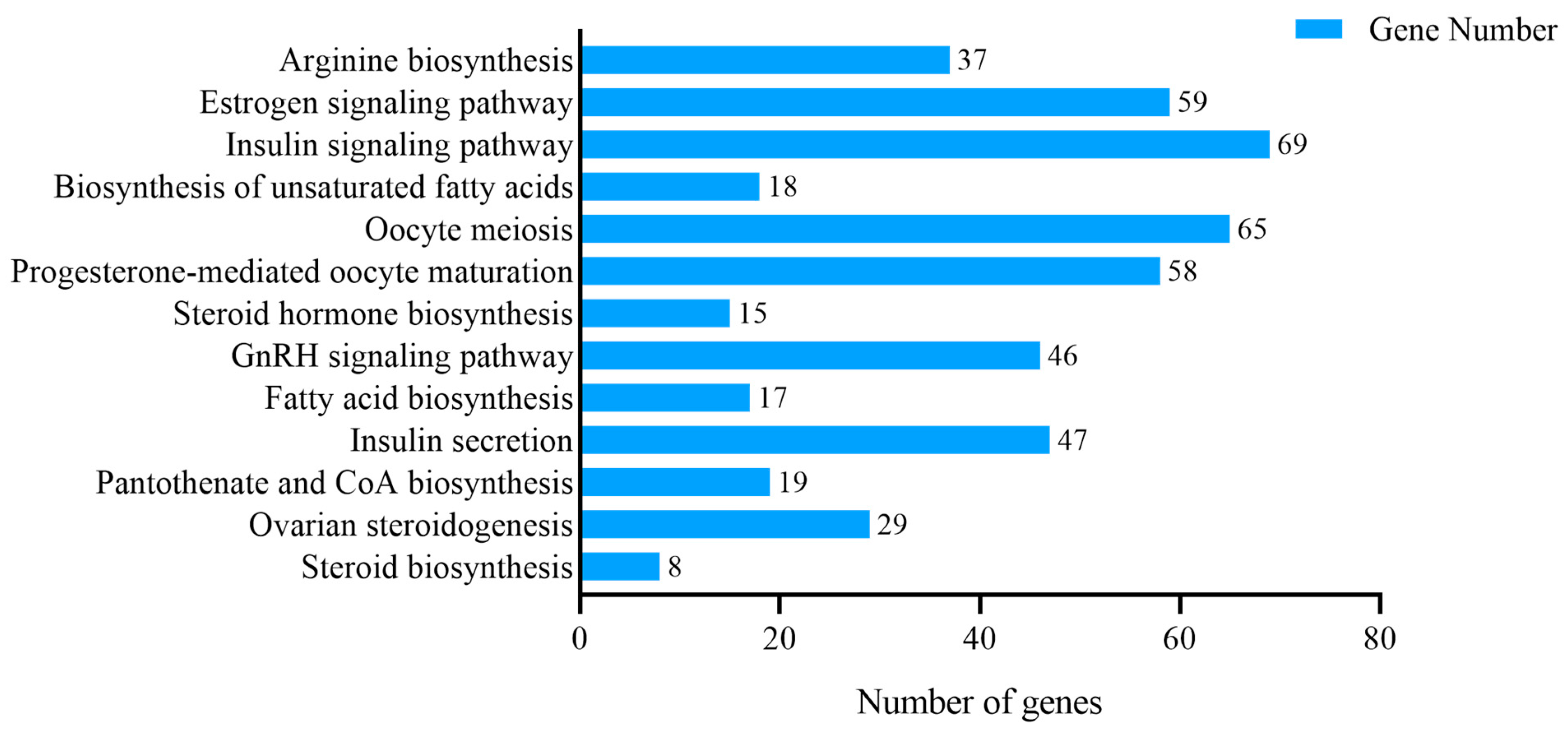
3.4. KEGG Enrichment Analysis
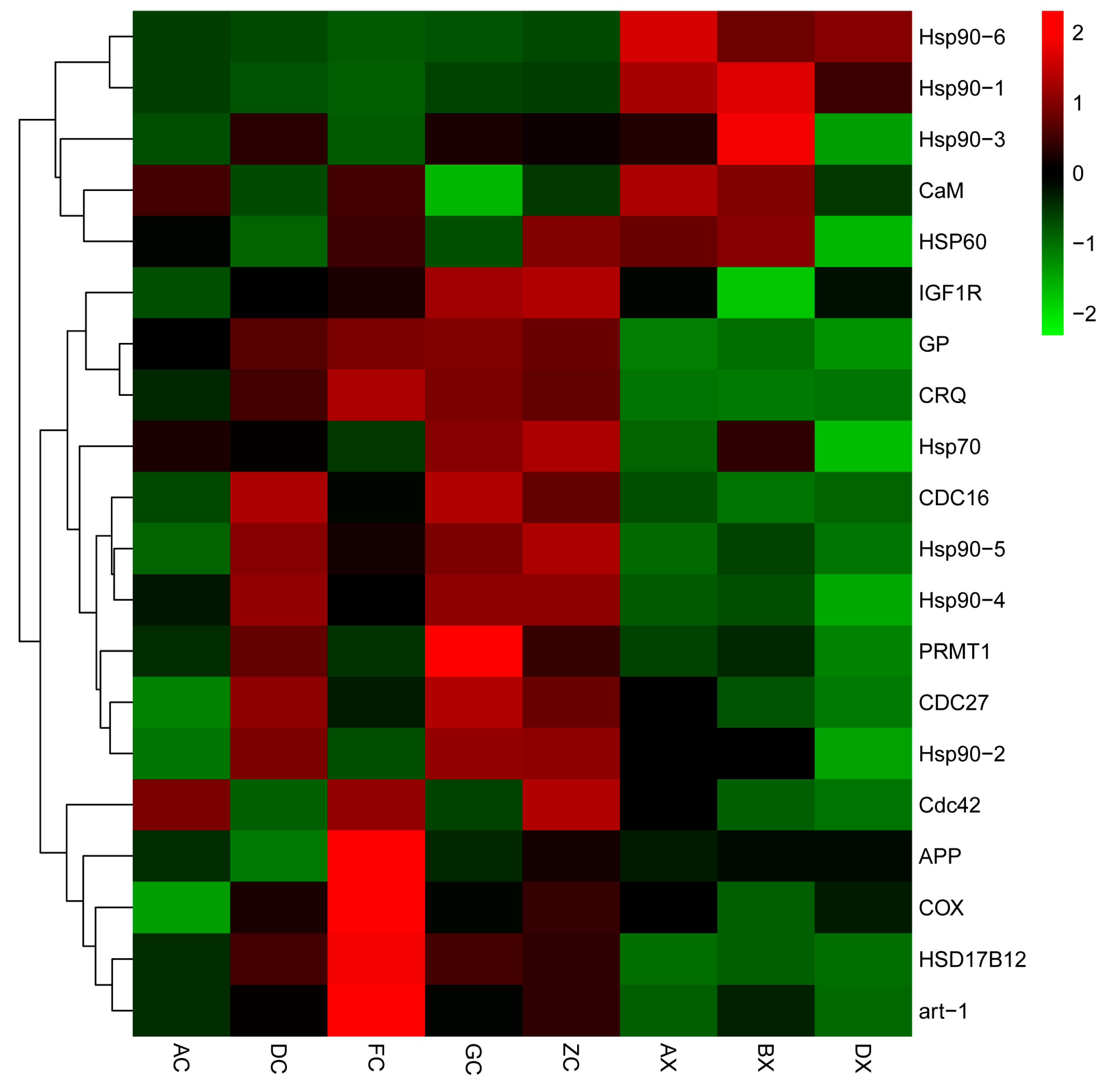
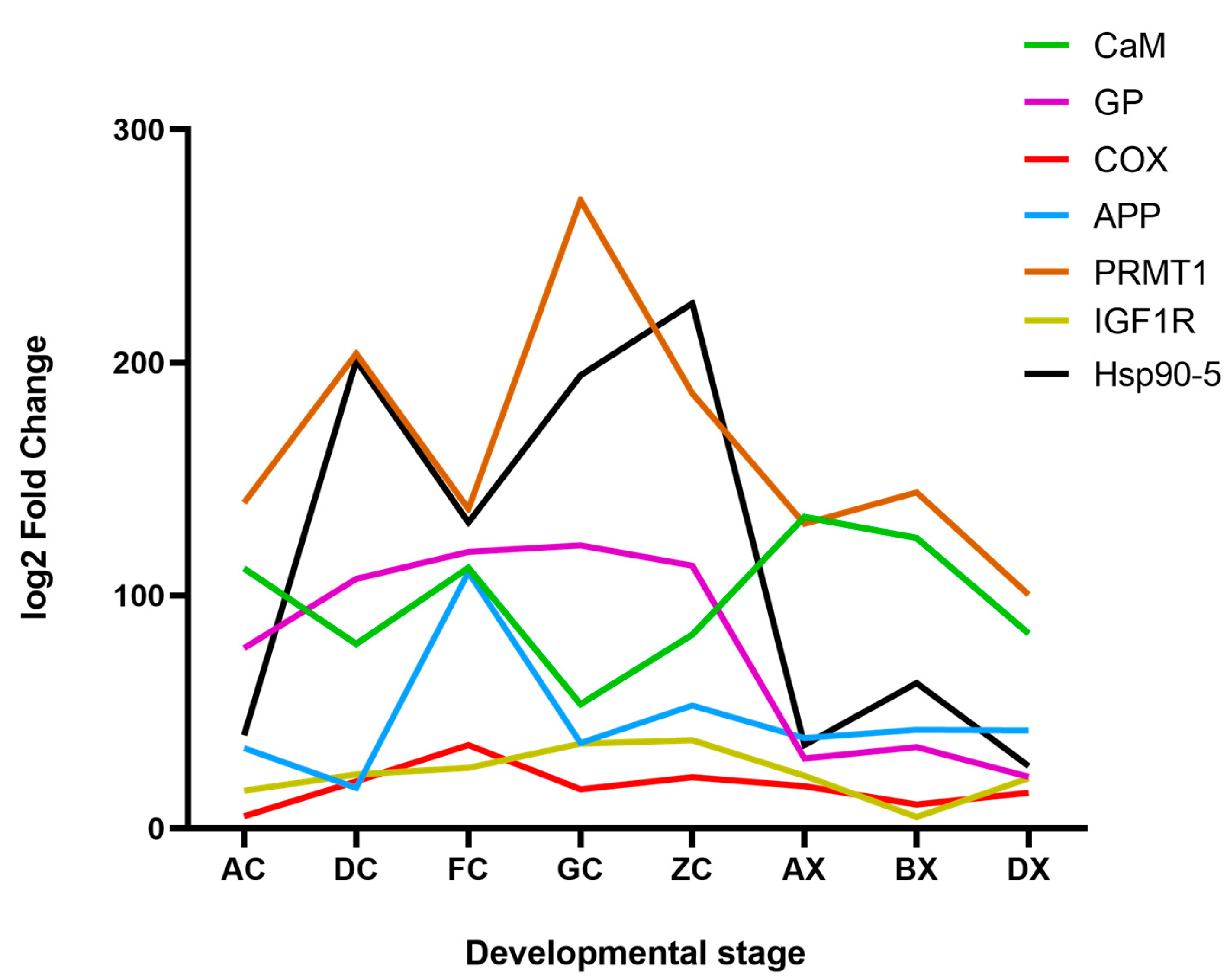
3.5. RT-qPCR Validation
4. Discussion
4.1. Analysis of Gonadal Development-Related Pathways
4.2. Key Differentially Expressed Genes Related to Gonadal Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fao, F.; Bondad-Reantaso, M.G.; Arthur, J.R. FAO Fisheries and Aquaculture Report NFIA/R1333 (En); FAO: Rome, Italy, 2021. [Google Scholar]
- Zhou, J.S.; Cao, Z.M.; Yang, G.L.; Li, J.L.; Zhang, Z.W.; Wu, T.T. Study on hybrid of burma introduced and Zhejiang locally-cultured populations of giant prawn Macrobrachium rosenbergii de man and SRAP marker. J. Fish. Sci. China 2006, 13, 667–673. [Google Scholar]
- Luo, K.; Yang, G.L.; Kong, J.; Zhang, Y.F.; Wang, J.Y.; Gong, J.H.; Gao, Q. Analysis of the effect of hybridization between different groups of Macrobrachium rosenbergii. Mar. Fish. Res. 2008, 29, 67–73. [Google Scholar]
- Zhao, Y.L.; Peng, X.X.; Li, X. Histological study on the female reproductive system of Macrobrachium rosenbergii. J. East China Norm. Univ. (Nat. Sci.) 1998, 3, 81–85. [Google Scholar]
- Wang, Y.F.; Du, N.S.; Lai, W. Histochemical tudy on the male reproductive system of Macrobrachium rosenbergii. J. Fish. Sci. China 1997, 4, 14–17. [Google Scholar]
- Zhu, Y.K.; Chen, H.L.; Wei, Y.F.; Wang, W.M. Effects of Mrfoxl 2-dsRNA on the expression of foxl 2 and reproduction-related genes in the ovariesof Macrobrachium rosenbergii. Freshw. Fish. 2022, 52, 38–43. [Google Scholar] [CrossRef]
- Chen, X.F.; Wang, C.L.; Gu, Z.M.; Xu, B.P.; Zhang, Y.F.; Shen, P.J.; Cheng, H.H.; Peng, F.; Li, X.L.; Huang, Z.Y. Transcriptome analysis of Macrobrachium rosenbergii ovary in four development stages. Oceanol. Et Limnol. Sin. 2019, 50, 398–408. [Google Scholar]
- Jia, C.L.; Zhang, Y.; Zhu, L.; Zhang, R. Application progress of transcriptome sequencing technology in biological sequencing. Mol. Plant Breed. 2015, 13, 2388–2394. [Google Scholar] [CrossRef]
- Wei, P.Y. Transcriptome and Identification of Differently Experesing Genes in the Testis and Ovary of Pacific White Shrimp LitopenaeusI Vannamei Broodstock. Master’s Thesis, Guangxi University, Nanning, China, 2017. [Google Scholar]
- Jiang, J.; Yuan, X.; Qiu, Q.; Huang, G.; Jiang, Q.; Fu, P.; Zhang, Y.; Jia, Y.; Yang, X.; Jiang, H. Comparative transcriptome analysis of gonads for the identification of sex-related genes in giant freshwater prawns (Macrobrachium rosenbergii) using RNA sequencing. Genes 2019, 10, 1035. [Google Scholar] [CrossRef]
- Jiang, H.X. Screening, Cloning, Expression and Functional Analysis of Sexual Precocity Related Genes in Female Macrobrachium Nipponense. Ph.D. Thesis, North West Agriculture and Forestry University, Xianyang, China, 2017. [Google Scholar]
- Ci, Y.J. Preliminary Study on Molecular Mechanism of Precious Chinese Mitten Crab. Master’s Thesis, Shanghai Ocean University, Shanghai, China, 2015. [Google Scholar]
- Chen, D.M. Ontogenetic Development of Gonads and Transcriptomeanalysis of the Peppermint Shrimp, Lysmata vittata. Master’s Thesis, Xiamen University, Xiamen, China, 2019. [Google Scholar]
- Waiho, K.; Fazhan, H.; Shahreza, M.S.; Moh, J.H.Z.; Noorbaiduri, S.; Wong, L.L.; Sinnasamy, S.; Ikhwanuddin, M. Transcriptome analysis and differential gene expression on the testis of orange mud crab, Scylla olivacea, during sexual maturation. PLoS ONE 2017, 12, e0171095. [Google Scholar] [CrossRef]
- Chen, G.Z.; Zhong, Z.X.; Yang, M.M.; Xing, Q.Q.; Xia, Z.L.; Tang, Q.Y.; Yi, S.K.; Cai, M.Y.; Yang, J.; Yang, G.L. Histological observation of gonad development during the first sexual maturity of Macrobrachium rosenbergii. Oceanol. Et Limnol. Sin. 2022, 53, 1208–1218. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Deveraux, Q.; Turk, B.; Sali, A. Comprehensive search for cysteine cathepsins in the human genome. Biol. Chem. 2004, 385, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Mario, S.; Burkhard, M. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar]
- Jaina, M.; Finn, R.D.; Eddy, S.R.; Alex, B.; Marco, P. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 11, R106. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Y.; Wang, N.; Wang, C.A.; Qu, S. Biological Functions of Arginine. Feed Res. 2009, 2, 16–18. [Google Scholar]
- Baños, N.; Planas, J.V.; Gutiérrez, J.; Navarro, I. Regulation of plasma insulin-like growth factor-I levels in brown trout (Salmo trutta). Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1999, 124, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Jiao, B.; Huang, X.; Chan, C.B.; Zhang, L.; Wang, D.; Cheng, C.H.K. The co-existence of two growth hormone receptors in teleost fish and their differential signal transduction, tissue distribution and hormonal regulation of expression in seabream. J. Mol. Endocrinol. 2006, 36, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-S.; Jiao, B.; Hu, C.; Huang, X.; Liu, Z.; Cheng, C.H.K. Discovery of a gonad-specific IGF subtype in teleost. Biochem. Biophys. Res. Commun. 2008, 367, 336–341. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Ding, L.; Zhang, Y.; Wang, Y.; Zhang, B.S. Regulation of Insulin in Secretion and Activity of Gonadotropin-Releasing Hormone. Chin. J. Anim. Nutr. 2013, 25, 1–7. [Google Scholar]
- DiVall, S.A.; Radovick, S.; Wolfe, A. Egr-1 binds the GnRH promoter to mediate the increase in gene expression by insulin. Mol. Cell. Endocrinol. 2007, 270, 64–72. [Google Scholar] [CrossRef]
- Liu, F.; Usui, I.; Evans, L.G.; Austin, D.A.; Mellon, P.L.; Olefsky, J.M.; Webster, N.J. Involvement of both G(q/11) and G(s) proteins in gonadotropin-releasing hormone receptor-mediated signaling in L beta T2 cells. J. Biol. Chem. 2002, 277, 32099–32108. [Google Scholar] [CrossRef] [PubMed]
- Navratil, A.M.; Song, H.; Hernandez, J.B.; Cherrington, B.D.; Santos, S.J.; Low, J.M.; Do, M.-H.T.; Lawson, M.A. Insulin augments gonadotropin-releasing hormone induction of translation in LβT2 cells. Mol. Cell. Endocrinol. 2009, 311, 47–54. [Google Scholar] [CrossRef]
- Xu, J.; Kirigiti, M.A.; Grove, K.L.; Smith, M.S. Regulation of Food Intake and Gonadotropin-Releasing Hormone/Luteinizing Hormone during Lactation: Role of Insulin and Leptin. Endocrinology 2009, 150, 4231–4240. [Google Scholar] [CrossRef] [Green Version]
- Soonthornsumrith, B.; Saetan, J.; Kruangkum, T.; Thongbuakaew, T.; Senarai, T.; Palasoon, R.; Sobhon, P.; Sretarugsa, P. Three-dimensional organization of the brain and distribution of serotonin in the brain and ovary, and its effects on ovarian steroidogenesis in the giant freshwater prawn, Macrobrachium rosenbergii. Invertebr. Neurosci. 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.L.; Tan, Y.J.; Wu, J.M.; Yan, X.Z.; Bao, X.S.; Yao, J.M. Changes of Hemolymph 20-hydroxyecdysone 17β-oestradiol and Testosteronelevels in Eriocheir Sinensis. J. Fish. China. 1992, 16, 101–106. [Google Scholar]
- Chandler, J.C.; Aizen, J.; Elizur, A.; Hollander-Cohen, L.; Battaglene, S.C.; Ventura, T. Discovery of a novel insulin-like peptide and insulin binding proteins in the Eastern rock lobster Sagmariasus verreauxi. Gen. Comp. Endocrinol. 2015, 215, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Dircksen, H.; Neupert, S.; Predel, R.; Verleyen, P.; Huybrechts, J.; Strauss, J.; Hauser, F.; Stafflinger, E.; Schneider, M.; Pauwels, K.; et al. Genomics, Transcriptomics, and Peptidomics of Daphnia pulex Neuropeptides and Protein Hormones. J. Proteome Res. 2011, 10, 4478–4504. [Google Scholar] [CrossRef]
- Reinecke, M.; Björnsson, B.T.; Dickhoff, W.W.; McCormick, S.D.; Navarro, I.; Power, D.M.; Gutiérrez, J. Growth hormone and insulin-like growth factors in fish: Where we are and where to go. Gen. Comp. Endocrinol. 2005, 142, 20–24. [Google Scholar] [CrossRef]
- Duleba, A.J.; Spaczynski, R.Z.; Olive, D.L. Insulin and insulin-like growth factor I stimulate the proliferation of human ovarian theca-interstitial cells. Fertil. Steril. 1998, 69, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Brogiolo, W.; Stocker, H.; Ikeya, T.; Rintelen, F.; Fernandez, R.; Hafen, E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 2001, 11, 213–221. [Google Scholar] [CrossRef]
- Benninghoff, A.D.; Thomas, P. Involvement of calcium and calmodulin in the regulation of ovarian steroidogenesis in Atlantic croaker (Micropogonias undulatus) and modulation by Aroclor 1254. Gen. Comp. Endocrinol. 2005, 144, 211–223. [Google Scholar] [CrossRef]
- Wang, A.X. Calmodulin and Sperm Function. Int. J. Urol. Nephrol. 1996, 4, 177–179. [Google Scholar]
- Örn, S.; Yamani, S.; Norrgren, L. Comparison of Vitellogenin Induction, Sex Ratio, and Gonad Morphology between Zebrafish and Japanese Medaka after Exposure to 17α-Ethinylestradiol and 17β-Trenbolone. Arch. Environ. Contam. Toxicol. 2006, 51, 237–243. [Google Scholar] [CrossRef]
- Brown, M.A.; Zhu, L.; Schmidt, C.; Tucker, P.W. Hsp90—From signal transduction to cell transformation. Biochem. Biophys. Res. Commun. 2007, 363, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y. Expression and Function Analysis of Porcine PRMTI Gene. Master’s Thesis, Xinyang Normal University, Xinyang, China, 2017. [Google Scholar]
- Jiang, X. Molecular Cloning, Characterization and Expression Analysis of Proteinarginine N-methyltransferase 1 gene (As-PRMTI) from Artemia sinica. Master’s Thesis, Liaoning Normal University, Dalian, China, 2015. [Google Scholar]
- Yan, H.; Cui, X.; Shen, X.; Wang, L.; Jiang, L.; Liu, H.; Liu, Y.; Liu, Q.; Jiang, C. De novo transcriptome analysis and differentially expressed genes in the ovary and testis of the Japanese mantis shrimp Oratosquilla oratoria by RNA-Seq. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 26, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.B.; You, J.G.; Li, X.; Yong, G. Research progress of prostaglandins in aquatic animals. Jiangxi Fish. Sci. Technol. 2019, 168, 41–43. [Google Scholar]
- Sreenivasula Reddy, P.; Ramachandra Reddy, P.; Purna Chandra Nagaraju, G. The synthesis and effects of prostaglandins on the ovary of the crab Oziotelphusa senex senex. Gen. Comp. Endocrinol. 2004, 135, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Tahara, D.; Yano, I. Maturation-related variations in prostaglandin and fatty acid content of ovary in the kuruma prawn (Marsupenaeus japonicus). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2004, 137, 631–637. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Gene Name | Sequence 5′-3′ | Length (bp) |
|---|---|---|---|
| 1 | CRQ Croquet protein | F: CCGCAGAACGCAAAGAGAAT | 228 |
| R: TGACAGTTGGACAGCAGACA | 214 | ||
| 2 | COX Cyclooxygenase | F: TCATCCGTGGCAGTTCTTGT | 251 |
| R: GCACTTGTGGCATCTGGTATC | 224 | ||
| 3 | APP Amyloid-beta-like protein | F: GATGATGACGACGACGATGAC | 201 |
| R: TCAGGTGGAGTAGAGGCATTG | 283 | ||
| 4 | Cdc42 Cell division control protein 42 | F: CAGACCATCAAGTGCGTAGTG | 256 |
| R: CACGGAGAAGCAGACCAGAA | 201 | ||
| 5 | Hsd17b12 Hydroxysteroid (17-beta) dehydrogenase 12a | F: CCTACACTTATGGCTCCAACAC | 249 |
| R: CTCCCTCTCCTTCCTCTTCAAA | 234 | ||
| 6 | Art-1 CoA reductase-1 | F: CGTTGCTTACCATGTGAACCA | 262 |
| R: TCCAAGAACCAACCTCGTATGT | 249 | ||
| 7 | Hsp70 Heat shock protein 70 | F: CCAAGCAGACTCAGACATTCAC | 216 |
| R: CAGCAGACACATTCAGGATACC | 282 | ||
| 8 | Hsp90 Heat shock protein 90 | F: AACTGTCTCGCTCCACTTGA | 228 |
| R: TACCTTCCTCCTCATCTTCCTC | 239 | ||
| 9 | PRMT1 Protein arginine methyltransferases 1 | F: TTCTCCTCTACGCCGCTTT | 200 |
| R: GTTCCGCAACCAACATCCA | 224 | ||
| 10 | GP Glycogen phosphorylase | F: GCTCAACGACACTCATCCTTC | 207 |
| R: CGCCTGCTAACTTCCTGTAGA | 203 | ||
| 11 | 18S rRNA | F: TATACGCTAGTGGAGCTGGAA | 286 |
| R: GGGGAGGTAGTGACGAAAAAT | 257 |
| Sample ID | Tissue | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| AC1 | STⅠ ovary | 22,801,902 | 22,362,483 | 6.71G | 0.02 | 98.1 | 94.37 | 41.95 |
| AC2 | STⅠ ovary | 23,113,581 | 22,616,983 | 6.79G | 0.02 | 98.18 | 94.43 | 41.41 |
| AC3 | STⅠ ovary | 20,423,838 | 20,034,059 | 6.01G | 0.03 | 98.06 | 94.04 | 41.75 |
| DC1 | STⅢ ovary | 23,366,938 | 22,945,237 | 6.88G | 0.02 | 98.22 | 94.4 | 41.51 |
| DC2 | STⅢ ovary | 22,622,979 | 22,155,554 | 6.65G | 0.02 | 98.4 | 94.89 | 41.64 |
| DC3 | STⅢ ovary | 20,350,586 | 19,954,445 | 5.99G | 0.02 | 98.25 | 94.48 | 41.34 |
| FC1 | STⅥ ovary | 22,058,708 | 21,518,653 | 6.46G | 0.03 | 98.06 | 94.11 | 41.23 |
| FC2 | STⅥ ovary | 21,638,453 | 21,203,487 | 6.36G | 0.02 | 98.17 | 94.29 | 41.17 |
| FC3 | STⅥ ovary | 23,790,503 | 23,267,866 | 6.98G | 0.02 | 98.38 | 94.78 | 41.51 |
| GC1 | STⅦ ovary | 21,968,333 | 21,489,585 | 6.45G | 0.02 | 98.32 | 94.64 | 40.99 |
| GC2 | STⅦ ovary | 24,898,145 | 24,301,643 | 7.29G | 0.02 | 98.34 | 94.73 | 41.98 |
| GC3 | STⅦ ovary | 22,953,131 | 22,439,268 | 6.73G | 0.02 | 98.17 | 94.34 | 41.84 |
| ZC1 | STⅧ ovary | 20,212,619 | 19,796,150 | 5.94G | 0.02 | 98.29 | 94.58 | 41.68 |
| ZC2 | STⅧ ovary | 22,706,643 | 22,141,590 | 6.64G | 0.02 | 98.3 | 94.71 | 41.93 |
| ZC3 | STⅧ ovary | 23,083,488 | 22,496,884 | 6.75G | 0.02 | 98.24 | 94.57 | 41.68 |
| AX1 | STⅠ testis | 23,730,986 | 23,280,534 | 6.98G | 0.02 | 98.25 | 94.54 | 40.55 |
| AX2 | STⅠ testis | 22,838,934 | 22,222,520 | 6.67G | 0.02 | 98.23 | 94.52 | 41.11 |
| AX3 | STⅠ testis | 23,593,669 | 23,074,458 | 6.92G | 0.02 | 98.37 | 94.81 | 40.24 |
| BX1 | STⅡ testis | 23,037,584 | 22,343,012 | 6.70G | 0.02 | 98.27 | 94.61 | 42.25 |
| BX2 | STⅡ testis | 22,223,635 | 21,787,130 | 6.54G | 0.02 | 98.21 | 94.34 | 39.27 |
| BX3 | STⅡ testis | 22,571,163 | 22,255,874 | 6.68G | 0.02 | 98.12 | 94.06 | 41.27 |
| DX1 | STⅢ testis | 23,243,749 | 22,870,217 | 6.86G | 0.02 | 98.18 | 94.26 | 39.77 |
| DX2 | STⅢ testis | 22,337,164 | 21,865,987 | 6.56G | 0.02 | 98.24 | 94.54 | 41.8 |
| DX3 | STⅢ testis | 22,273,017 | 21,797,186 | 6.54G | 0.02 | 98.19 | 94.44 | 42.53 |
| Database for Annotation | Number of Unigenes | Percentage of Annotated Unigenes (%) |
|---|---|---|
| Annotated in Nr | 27,684 | 18.89 |
| Annotated in Nt | 7474 | 5.10 |
| Annotated in KO | 8781 | 5.99 |
| Annotated in SwissProt | 15,032 | 10.25 |
| Annotated in Pfam | 30,530 | 20.83 |
| Annotated in GO | 30,520 | 20.82 |
| Annotated in KOG | 7278 | 4.96 |
| Annotated in all databases | 2148 | 1.46 |
| Annotated in at least one database | 43,427 | 29.63 |
| Total unigenes | 146,537 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, Z.; Chen, G.; Tu, H.; Yao, X.; Peng, X.; Lan, X.; Tang, Q.; Yi, S.; Xia, Z.; Cai, M.; et al. Transcriptomic Analysis and Functional Gene Expression in Different Stages of Gonadal Development of Macrobrachium rosenbergii. Fishes 2023, 8, 94. https://doi.org/10.3390/fishes8020094
Zhong Z, Chen G, Tu H, Yao X, Peng X, Lan X, Tang Q, Yi S, Xia Z, Cai M, et al. Transcriptomic Analysis and Functional Gene Expression in Different Stages of Gonadal Development of Macrobrachium rosenbergii. Fishes. 2023; 8(2):94. https://doi.org/10.3390/fishes8020094
Chicago/Turabian StyleZhong, Zhenxiao, Guozhu Chen, Haihui Tu, Xinyi Yao, Xin Peng, Xuan Lan, Qiongying Tang, Shaokui Yi, Zhenglong Xia, Miaoying Cai, and et al. 2023. "Transcriptomic Analysis and Functional Gene Expression in Different Stages of Gonadal Development of Macrobrachium rosenbergii" Fishes 8, no. 2: 94. https://doi.org/10.3390/fishes8020094
APA StyleZhong, Z., Chen, G., Tu, H., Yao, X., Peng, X., Lan, X., Tang, Q., Yi, S., Xia, Z., Cai, M., & Yang, G. (2023). Transcriptomic Analysis and Functional Gene Expression in Different Stages of Gonadal Development of Macrobrachium rosenbergii. Fishes, 8(2), 94. https://doi.org/10.3390/fishes8020094