3.2. Reduction to TBRIM Like Case and Its Analysis
Using the methods described in [
48], we numerically compute a certain number of one-particle or orbital energy eigenvalues
and corresponding eigenstates
of the Sinai oscillator (
2). As repulsive interaction potential
we choose the short ranged box function
if
(since
) and
, otherwise. Here, the parameter
gives the overall scale of the interaction strength depending on the charge of the particles and eventually other physical parameters.
Therefore, the corresponding many-body Hamiltonian with
M one-particle orbitals and
spinless fermions takes the form:
where, for
and
, we have the interaction matrix elements:
and
,
are fermion operators for the
M orbitals satisfying the usual anticommutation relations. We note that, in the literature, when expressing a two-body interaction potential in second quantization, one usually uses the raw matrix elements
with an additional prefactor of
and full independent sums for the four indices
i,
j,
k and
l. Using the particle exchange symmetry:
, one can reduce the
sums to
, which removes the prefactor
(after exchanging the index names
l and
k for the
contributions and exploiting that contributions at
or
obviously vanish). The definition of the anti-symmetrized interaction matrix elements
according to (
5) allows for also reducing the
sums to
. Furthermore, the ordering of the two fermion operators
in (
4) is also important and necessary to obtain positive expectation values if the interaction is repulsive. The anti-symmetrized matrix elements
correspond to a
matrix with
. In order to avoid a global shift of the non-interacting eigenvalue spectrum due to the interaction, we also apply a diagonal shift
to ensure that this matrix has a vanishing trace ( One can easily show that the trace of the
anti-symmetrized interaction matrix is proportional to the trace of the interaction operator in the many-body Hilbert space with a factor depending on
M and
L). Of course, such a global energy shift does not affect the issues of thermalization, interaction induced eigenfunction mixing or the quantum time evolution with respect to the Hamiltonian
H, etc.
We note that the transition from the classical Hamiltonian (
2) to the quantum one (
4) is done by the standard procedure of second quantization (see, e.g., [
68]).
3.3. Aberg Parameter
In absence of interaction, the energy eigenvalues of (
4) are given as the sum of occupied orbital energies:
where
represents a configuration such that
and
. The associated eigenstates are the basis states where each orbital is either occupied (if
) or unoccupied (if
) and, in this work, we will denote these states in the usual occupation number representation:
, where, for convenience, we write the lower index orbitals starting from the right side.
The distribution of the total one-particle energies (
6) is numerically rather close to a Gaussian (since
act as quasi-random numbers) with mean and variance (see also Equation (A.4) of Ref. [
46]):
Therefore, the many-body level spacing
or inverse Heisenberg time at the band center
is given by
where
is the dimension of the fermion Hilbert space in the sector of
M orbitals and
L particles. In our numerical computations, we simply evaluated the quantities
of (
7) using the exact one-particle energy eigenvalues obtained from the numerical diagonalization of the one-particle Sinai Hamiltonian
given in (
2). However, to get some analytical simplification for large
M, one may use the one-particle density of states (
3), which gives, after replacing the sums by integrals and neglecting the constant term,
and
.
For the question of whether the interaction strength is sufficiently strong to mix the non-interacting basis states, the important quantity is the effective level spacing of states coupled directly by the interaction
where
is the number of nonzero elements for a column (or row) of
H [
41,
69] and we need to use the variance for only two particles:
because the interaction only couples states where (at least)
particles are on the same orbital such that (at most) only the partial sum of two one-particle energies is different between two coupled states. Even though for two particles the hypothesis of a Gaussian distribution is theoretically not justified, the distribution is still sufficiently similar to a Gaussian and it turns out that the value of
as the coupled two-particle density of states in the band center is numerically quite accurate with an error below 10% (for
and our choice of
values).
According to the Åberg criterion [
38,
39,
41], the onset of chaotic mixing happens for typical interaction matrix elements
U comparable to
. Therefore, we compute the quantity
(which is proportional to the interaction amplitude
U) where the average is done with respect to all
matrix elements of the interaction matrix. This quantity might be problematic and not correspond to a typical interaction matrix element in the case of a long tail distribution. However, in our case, it turns out that
, which excludes this scenario. Using this quantity, we introduce the dimensionless Åberg parameter and the critical interaction amplitude
by
such that
if
. We expect [
38,
39,
41] to be the onset of strong/chaotic mixing at
and a perturbative regime for
, while, at
, we have the critical interaction strength
. The value of
depends on the parameters
L,
M,
and the overlap of the one-particle eigenstates according to (
5). To obtain some useful analytical expression of
, we note that the quantity
, numerically computed for
, can be quite accurately fitted by
. Furthermore, we remind readers of the expression
which can be simplified in the limit
and
, such that
, resulting in:
. Here, we also used the found expression above
. From this, we find that
with a numerical constant
, where we also used
according to (
3). Below, we will give more accurate numerical values of
,
and
for the parameter choice of
M and
L numerically relevant in this work.
We note that this estimate for
applies to energies close to the many body band center of
H and that, for energies away from the band center, the value of
is enhanced, thus reducing the effective value of
A. Furthermore, we computed
by a simplified average over
all interacting matrix elements not taking into account an eventual energy dependence according to the index values of
in (
5).
3.4. Density of States
In this work, we present numerical results for the case of
orbitals and
particles corresponding to a many-body Hilbert space of dimension
and the number
of directly coupled states of a given initial state by non-vanishing interaction matrix elements in (
4). Thus, in our studies, the whole Hilbert space is built only on these
orbitals. We diagonalize numerically the many-body Hamiltonian (
4) for various values of
A in the range
. We have also verified that the results and their physical interpretation are similar for smaller cases such as
,
(with
,
) or
,
(
,
).
We mention that, for
and
, we find numerically that
and, from (
8), that
, where the quantities
were exactly computed from the numerical orbitals energies
. From this, we find that
. This expression is more accurate than the more general analytical estimate for arbitrary
and
given in the last section (which would provide
for
and
).
Our first observation is that, even in the presence of interactions, the density of states has approximately a Gaussian form with the same center
given in (
7) for the case
. This is simply due the fact that the interaction matrix has, by choice, a vanishing trace and does not provide a global shift of the spectrum. We determine the variance
of the Gaussian density of states by a fit of the integrated density of states
using
such that
is a Gaussian of width
and center
(see Appendix A of Ref. [
46] for more details). From this, we find the behavior:
where
is a constant depending on
M and
L; for
,
the fit values of
and
are
and
. It is also possible to determine
using the expression
where the trace in Fock space can be evaluated either by using the matrix
H before diagonalizing it or using its exact energy eigenvalues
. This provides the same behavior as (
10) with the very similar numerical values
and
(for
,
). We mention that the integrated Gaussian density of states (
9) is not absolutely exact but quite accurate for values
. For larger values of
A, the deviations increase, but the overall form is still correct. As described in [
46], the quality of the fit can be considerably improved if we replace in (
9) the linear function
by a polynomial of degree 5. In this case, the precision of the fit is highly accurate for the full range of
A values we consider. In particular, we use this improved fit to perform the spectral unfolding when computing the nearest level spacing distribution (shown below).
To obtain some theoretical understanding of (
10), one can consider a model where the initial interaction matrix elements (
5) are replaced by independent Gaussian variables with identical variance
. In this case, one can show theoretically [
46] that
, where
is a number somewhat larger than
K taking into account that certain non-vanishing interaction matrix elements in Fock space are given as a sum of
several initial interaction matrix elements (
5) (see Appendix A of [
46] for details). The parameter
takes for
,
(
,
or
,
) the value
(
or
, respectively). Since
we indeed obtain (
10) with
. For
,
, we find
(see (
7)) and
. The latter is roughly by a factor of 2 smaller than the numerical value. We attribute this to the fact that the real initial interaction matrix elements (
5) are quite correlated, and not independent uniform Gaussian variables, leading therefore to an effective increase of the number
due to hidden correlations. The important point is that theoretically at very large values values of
M and
L, e.g.,
, we have
and
, which is parametrically small for very large
L. Therefore, there is a considerable range of values
where the interaction strongly mixes the non-interacting many-body eigenstates but where the density of states is only weakly affected by the interaction. This regime is also known as the Breit–Wigner regime (see, e.g., [
40], for the case of interacting Fermi systems).
3.5. Thermalization and Entropy of Eigenstates
In the following, we mostly concentrate on values
such that the effect of the increase of the spectral width
is still small or at least quite moderate. The question arises if a given many-body state, either an exact eigenstate of
H or a state obtained from a time evolution with respect to
H, is thermalized according to the Fermi–Dirac distribution [
47]. As in [
45,
46], we determine the occupation numbers
for such a state, as well as the corresponding fermion entropy
S [
47] and the effective total one-particle energy
by:
based on the assumption of weakly interacting fermions. In the regime of modest interaction
(for
,
), corresponding to a constant spectral width
, we have typically
(for exact eigenstates of
H) or
(for other states). If the given state is thermalized, its occupation numbers
should be close to the theoretical Fermi–Dirac filling factor
with
, where inverse temperature
and chemical potential
are determined by the conditions:
Here,
E is normally given by
, but one may also consider the value
(or
) provided the latter is in the energy interval where the conditions (
12) allow for a unique solution. Furthermore, for a given energy
E, we can also determine the theoretical (or thermalized) entropy
using (
11) with
being replaced by
(where
,
are determined from (
12) for the energy
E).
The many-body states with energies above
are artificial since they correspond to negative temperatures due to the finite number of orbitals considered in our model. Therefore, we limit our studies to the lower half of the energy spectrum
(for
,
). In
Figure 2, we compare the thermalized Fermi–Dirac occupation number
with the occupation numbers
for two eigenstates at level numbers
(1354, with
corresponding to the ground state) with approximate energy eigenvalue
(
) for three different Åberg parameters
,
and
. These states are not too close to the ground state but still quite far below the band center.
We note that the regime of negative temperatures is natural for the TBRIM where the energy spectrum is inside a finite energy band (this regime has been discussed in [
45,
46]). However, for the Sinai oscillator, the energy spectrum is unbounded and, due to that, the regime of negative temperatures, appearing in the numerical simulations due to a finite number of one-particle orbital, is artificial.
At weak interaction,
, both states are not at all thermalized with occupation numbers being either close to 1 or 0. Apparently, these states result from weak perturbations of the non-interacting eigenstates
or
, where the
values are rounded to 1 (or 0) if
(
). For
, the values of
are a little bit farther away from the ideal values 0 or 1 as compared to
but still sufficiently close to be considered as perturbative. Apparently, the state
, which is lower in the spectrum (with larger effective two-body level spacing), is less affected by the interaction than the state 1354. In both cases, the entropy
S is quite below the thermalized entropy
(see
Table 1 for numerical values of entropies, energies, inverse temperature and chemical potential for the states shown in
Figure 2).
At intermediate interaction, , the occupation numbers are closer to the theoretical Fermi–Dirac values but still with considerable deviations. Here, both entropy values S are rather close to . The state 1354 seems to be better thermalized than the state , the latter having a slightly larger deviation between both entropy values. At stronger interaction, , both states are very well thermalized with a good matching of both entropy values (again with the state 1354 being a bit better thermalized than the state ) provided we use as reference energy to compute temperature and chemical potential. The temperature obtained from is too small because here the increase of is already quite strong and rather strongly deviates from . In addition, the value of using does not match S. Obviously, at stronger interaction values, it is necessary to use to test the thermalization hypothesis of a given state.
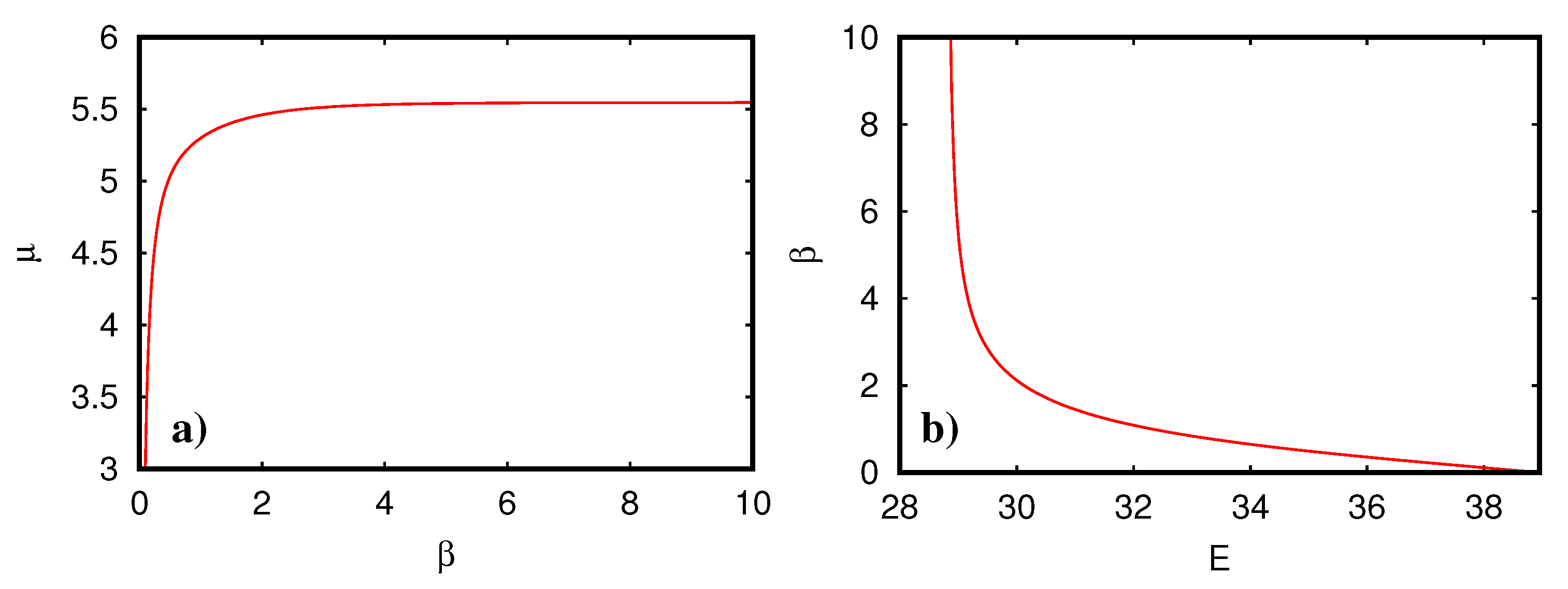
Figure 3 shows the mutual dependence between the three parameters
,
on
E when solving the conditions (
12). The chemical potential as a function of
is rather constant except for smallest values of
where
with a negative prefactor. One can actually easily show from (
12) that in the limit
the chemical potential does not depend on
and is given by
providing a singularity if
with negative (positive) prefactor for
(
) and
for
. The temperature (
) vanishes for
E close to the lower energy border and diverges for
E close to the band center
.
In
Figure 4, we present the nearest level spacing distribution
for different values of the Åberg parameter. To compute
, we have used only the “physical” levels in the lower half of the energy spectrum and the unfolding has been done with the integrated density of states (
9), where
is replaced by a fit polynomial of degree 5. For the smallest value
, the distribution
is very close to the Poisson distribution with some residual level repulsion at very small spacings. This is a quite well known effect because typically the transition from Wigner–Dyson to Poisson statistics (when tuning some suitable parameter such as the Åberg parameter from strong to weak coupling) is non-uniform in energy and happens first at larger spacings (energy differences) and then at smaller spacings. The reason is simply that two levels which by chance are initially very close are easily repelled by a small residual coupling matrix element (when slightly changing a disorder realization or similar). For
, there is somewhat more level repulsion at small spacings, but the distribution is still rather close to the Poisson distribution with some modest deviations for
. For the larger Åberg values
and
, we clearly obtain Wigner–Dyson statistics (taking into account the quite limited number of only
level spacing values for the histograms). These results clearly confirm that the transition from
to
corresponds indeed to a transition from a perturbative regime to a regime of chaotic mixing with Wigner–Dyson level statistics [
14].
A further confirmation that
is critical can be seen in
Figure 5, which compares the dependence of the entropy
S of exact eigenstates (lower half of the spectrum) on
or
with the theoretical thermalized entropy
. For the Åberg values
(and
), the entropy
S of all (most) states is significantly below its theoretical value
. Actually, the distribution of data points is considerably concentrated at smaller entropy values which is not so clearly visible in Figure. In particular, the average of the ratio of
is
for
and
for
. For the Åberg values
and
, most or nearly all entropy values (for
) are very close to the theoretical line with the average ratio
being
for
and
for
. For
, the states with lowest energies are not yet perfectly thermalized and the data points for
and
are still rather close. For
, all states are well thermalized (when using the energy
) while the data points for
are quite outside the theoretical curve simply due to the overall increase of the width of the energy spectrum. This observation is also in agreement with the discussion of
Figure 2. For smaller values
(not shown in
Figure 5), we find that the data points are still closer to the
E-axis while, for larger values
, the data points are clearly on the theoretical curve for
(but more concentrated on energy values closer to the center with larger entropy values and larger temperatures), while, for
, according to (
10), the overall width of the exact eigenvalue spectrum increases strongly and the data points are clearly outside the theoretical curve (except for a few states close to the band center).
We note that the data points in
Figure 5a,b significantly deviate from the theoretical thermalization curve since the Åberg criterion is not satisfied
. For
, the deviations are significantly reduced, but they are still more pronounced in a vicinity of the ground state in agreement with the relation (
1).
In
Figure 6, the occupation numbers
(averaged over several energy eigenvalues inside a given energy cell) are shown in the plane of energy
E and orbital index
k as color density plot for the Åberg parameter
. The comparison with the theoretical occupation numbers
(shown in the same way) provides further confirmation that, at
, there is indeed already a quite strong thermalization of most eigenstates.
3.6. Thermalization of Quantum Time Evolution
The question arises how or if a time dependent state
, obeying the quantum time evolution with the Hamiltonian
H and an initial state
being a non-interacting eigenstate
(with all
and
), evolves eventually into a thermalized state. We have computed such time dependent states using the exact eigenvalues and eigenvectors of
H to evaluate the time evolution operator. As initial states, we have chosen four states (for
,
): (i)
where a particle at orbital 7 is excited from the non-interacting ground state (with all orbitals from 1 to 7 occupied) to the orbital 12, (ii)
where two particles at orbitals 6 and 7 are excited from the non-interacting ground state to the orbitals 12 and 14, (iii)
and (iv)
. The states
and
are obtained from the exact eigenstate of
H for
at level number
and 1354, respectively, by rounding the occupation numbers
to 1 (or 0) if
(
) (states of top panels in
Figure 2). The approximate energies (
6) of these four states are
(
),
(
and
) and
(
).
It is useful to express the time in multiples of the elementary quantum time step defined as:
where
is the Heisenberg time (at the given value of
A),
d the dimension of the Hilbert space and
the width of the Gaussian density of states given in (
10). The quantity
is the shortest physical time scale of the system (inverse of the largest energy scale) and obviously for
the unitary evolution operator is close to the unit matrix multiplied by a uniform phase factor:
since the eigenvalues
of
H satisfy
. We expect that any signification deviation of
with respect to the initial condition
happens at
(or later in case of very weak interaction). Furthermore, by analyzing the time evolution in terms of the ratio
the results do not depend on the global energy scale of the spectral width. The longest time scale is the Heisenberg time
(since
for
,
). Later, we also discuss intermediate time scales such as the inverse decay rate obtained from the Fermi golden rule.
To show graphically the time evolution, we compute the time dependent occupation numbers
and present them in a color density plot in the plane
. In addition, at the time value used last, we compute the effective total one-particle energy
using the relation (
11) (note that
is not conserved with respect to the time evolution except for very weak interaction) and use this value to determine from (
12) the inverse temperature
, chemical potential
and the thermalized Fermi–Dirac filling factor
at each
k value for the orbital index. These values of ideally thermalized occupation numbers will be shown in an additional vertical bar ( Note that this additional bar is not related to the usual color bar that provides the translation of colors to
values). The latter is shown in
Figure 6 and applies also to all subsequent figures with color density plots for
values right behind the data for the last time values separated by a vertical white line. This presentation allows for an easy verification if the occupation numbers at the last time values are indeed thermalized or not.
In
Figure 7, we show the time evolution for the initial state
and the two Åberg parameter values
and
using a linear time scale with integer multiples of
and for
. At
, the occupation number
(of the excited particle) shows, at the beginning, a periodic structure, with an approximate period
for
, and a modest decay for
. At the same time, the first orbitals above the Fermi sea are slightly excited. At final
, the state is clearly not thermalized. For
, we see a very rapid partial decay of
and
together with an increase of
. Furthermore, for
with
, there are later and more modest excitations with a periodic time structure. Here, the final state at
is also not thermalized, but it is closer to thermalization as for the case
.
The linear time scale used in
Figure 7 is not very convenient since it cannot capture a rapid decay/increase of
well at small times and its maximal time value is also significantly limited below the Heisenberg time. Therefore, we use in
Figure 8 and
Figure 9 a logarithmic time scale with
. Note that, in these figures, the different
values for each cell are not time averaged but represent the precise values for certain, exponentially increasing, discrete time values (see caption of
Figure 8 for the precise values). Therefore, in case of periodic oscillations of
, there will be, for larger time values, a quasi random selection of different time positions with respect to the period.
In
Figure 8, the time evolution for the initial states
and
is shown for the Åberg values
. For
at
and
, the observations of
Figure 7 are confirmed with the further information that the absence of thermalization in these cases is also valid for time scales larger than
up to
and for
the initial decay of
and
happens at
. For
at
, the decay starts at
and an approximate thermalization happens at
. However, here there is still some time periodic structure and it would be necessary to do some time average to have perfect thermalization. For
at
, the decay of excited orbitals 12 and 14 starts at
and saturates at
at which time also orbitals 6 and 7 are excited. After this, there are very small excitations of orbitals 8, 9, 10 and maybe 13, 15. There is also some very modest decay of the Fermi sea orbitals 2, 4 and 5 at
. The final state at
is not thermalized even though some orbitals have
values close to thermalization. For
at
, the decay of excited orbitals 12 and 14 starts at
and, for
, there is thermalization (but requiring some time average as for
at
). Interestingly, at intermediate times
, the high orbitals 13 and 16 are temporarily slightly excited and decay afterwards rather quickly to their thermalized values. For
at
, the decay of excited orbitals 12 and 14 starts even at
and thermalization seems to set in at
.
Figure 9 is similar to
Figure 8 except for the initial states
and
which have occupation numbers
obtained by rounding the
values of the two eigenstates visible in the two top panels of
Figure 2. Here, the initial decay of excited orbitals starts roughly at
(
or
) for
(
or
, respectively). There is no thermalization for both states at
(but some
values are close to thermalized values), approximate thermalization for
and
and good thermalization for
and
as well as
(both states).
Using the time dependent values
, one can immediately determine the corresponding entropy
using (
11). At
, we have obviously
, since, for all four initially considered states, we have perfect occupation number values of either
or
. Naturally, one would expect that the entropy increases with a certain rate and saturates then at some maximal value which may correspond (or be lower) to the thermalized entropy
(with
determined for the state
at large times) depending if there is presence (or absence) of thermalization according to the different cases visible in
Figure 8 and
Figure 9. However, in the absence of thermalization, we see that there may also exist periodic oscillations with a finite amplitude at very long time scales.
In
Figure 10, we show the time dependent entropy
for the two initial states
,
and the three values
,
and
of the Åberg parameter. For
, there is indeed a rather rapid saturation of the entropy of both states at a maximal value that is indeed close to the thermalized entropy
. We note that
is not conserved at strong interactions and that its initial value
(
) at
evolves to
(
) at large times for
(
) corresponding roughly to
(
) visible as thin blue horizontal lines in
Figure 10. For
(or
), the thermalized entropy values, visible as thin green (red) lines, are lower as compared to the case
due to different final
values. For
and
, there is also saturation of
S to its thermalized value. For
and
, there seems to be an approximate saturation at a quite low value
but with periodic fluctuations in the range
. For
and
, there is a quite late and approximate saturation with some fluctuations that are visible for
and with
. For
and
, there is a late periodic regime for
with a quite large amplitude
and with
significantly below the thermalized entropy
. The panels using a normal (instead of logarithmic) time scale with
miss completely the long time limits for
and might incorrectly suggest that there is an early saturation at quite low values of
S.
The periodic (or quasi-periodic) time dependence of or , for the cases with lower values of A and/or an initial state with lower energy, indicates that, for such states, only a small number () of exact eigenstates of H contribute mostly in the expansion of in terms of these eigenstates.
Figure 10 also shows that the initial increase of
is rather comparable between the two states for identical values of
A even though the long time limit might be very different. Furthermore, a closer inspection of the data indicates that typically
is close to a quadratic behavior for
but which immediately becomes linear for
similarly as the transition probabilities between states in the context of time dependent perturbation theory. To study the approximate slope in the linear regime, we define (For practical reasons, we decide to incorporate the quantum time step
in the definition of
, i.e.,
is defined as the ratio of the initial slope
over the global spectral bandwidth
) the quantity
for
, where
is a numerical constant of order one. To determine
practically, we perform first the fit
for
and use the exponential decay rate
to perform a refined fit
for the interval
. From this, we determine
, which is rather close to
for
but not for larger values of
A where the decay time is reduced and not sufficiently large in comparison to the initial quadratic regime. Therefore, the quadratic term in the exponential is indeed necessary to obtain a reasonable fit quality. This procedure corresponds to an effective average of the value of
between 1 and roughly
, which is indeed useful to smear out some oscillations in the initial increase of
for smaller values
.
We note that, for many-body quantum systems, the exponential growth of entropy with time had been also discussed and numerically illustrated in [
40] (see also related publications in References 25 and 26 there). Recently, such an exponential growth of entropy has been discussed in [
62,
70].
Figure 11 shows the dependence of these values of
on the parameter
A for our four initial states. At first sight, one observes a behavior
for
and a saturation for larger values of
A. However, a more careful analysis shows that there are modest but clearly visible deviations with respect to the quadratic behavior in
A (power law fits
for
provide exponents close to
) and it turns out that these deviations correspond to a logarithmic correction:
with
(for fits with
) or with
(for fits with all
A values).
To understand this behavior, we write for sufficiently small times
(if
) or
(if
), where
is the small modification of
. Time dependent perturbation theory suggests that
for
and
for
such that still
with coefficients
dependent on
k (and also on
M,
L) and satisfying a linear relation to ensure the conservation of particle number. Using (
11) and neglecting corrections of order
, we obtain:
and
with
. Since
, we find indeed the behavior:
where
indicates an average over some modest values of
. The precise values of
may depend rather strongly on the orbital index
k and the initial state (see also
Figure 8 and
Figure 9), but the coefficients
,
depend only slightly on the initial state (see
Table 2). Furthermore, by replacing
to allow for a saturation at large
A and with a further fit parameter
, it is possible to describe the numerical data by the more general fit
for the full range of
A values.
3.7. Survival Probability and Fermi’s Golden Rule
The knowledge of the time dependent states allows us also to compute the decay function which represents the survival probability of the initial non-interacting eigenstate due to the influence of interactions. Again, for the very short time window , we expect a quadratic decay: with being the quantum expectation value with respect to and a numerical constant since represents roughly the spectral width of H. For , but such that , we have, according to Fermi’s golden rule: where is the decay rate (Again, for practical reasons and similarly to , we incorporate in the definition of the time scale , i.e., usual decay rate found in the literature and meaning that is defined as the ratio of the usual decay rate over the global spectral bandwidth) of the state.
To determine
numerically, we apply the fit:
in two steps. First, we use the interval
and, if
, corresponding to a rapid decay (which happens for larger values of
A), we repeat the fit for the reduced interval
. The choice of the Amplitude
and the condition
for the fit range allow for taking into account the effects due to the small initial window of quadratic decay. In
Figure 12, we show two examples for the initial state
and the Åberg values
and
. In both cases, the shown maximal time value
(if
) or
(if
) defines the maximal time value for the fit range. For
, the fit nicely captures the decay for
, while, for
, there are also some oscillations in the decay function for which the fit procedure is equivalent to some suitable average in the range
. For very small values of
A, the fit procedure also works correctly since it captures only the initial decay that is important if
does not decay completely at large times and which typically happens in the perturbative regime
.
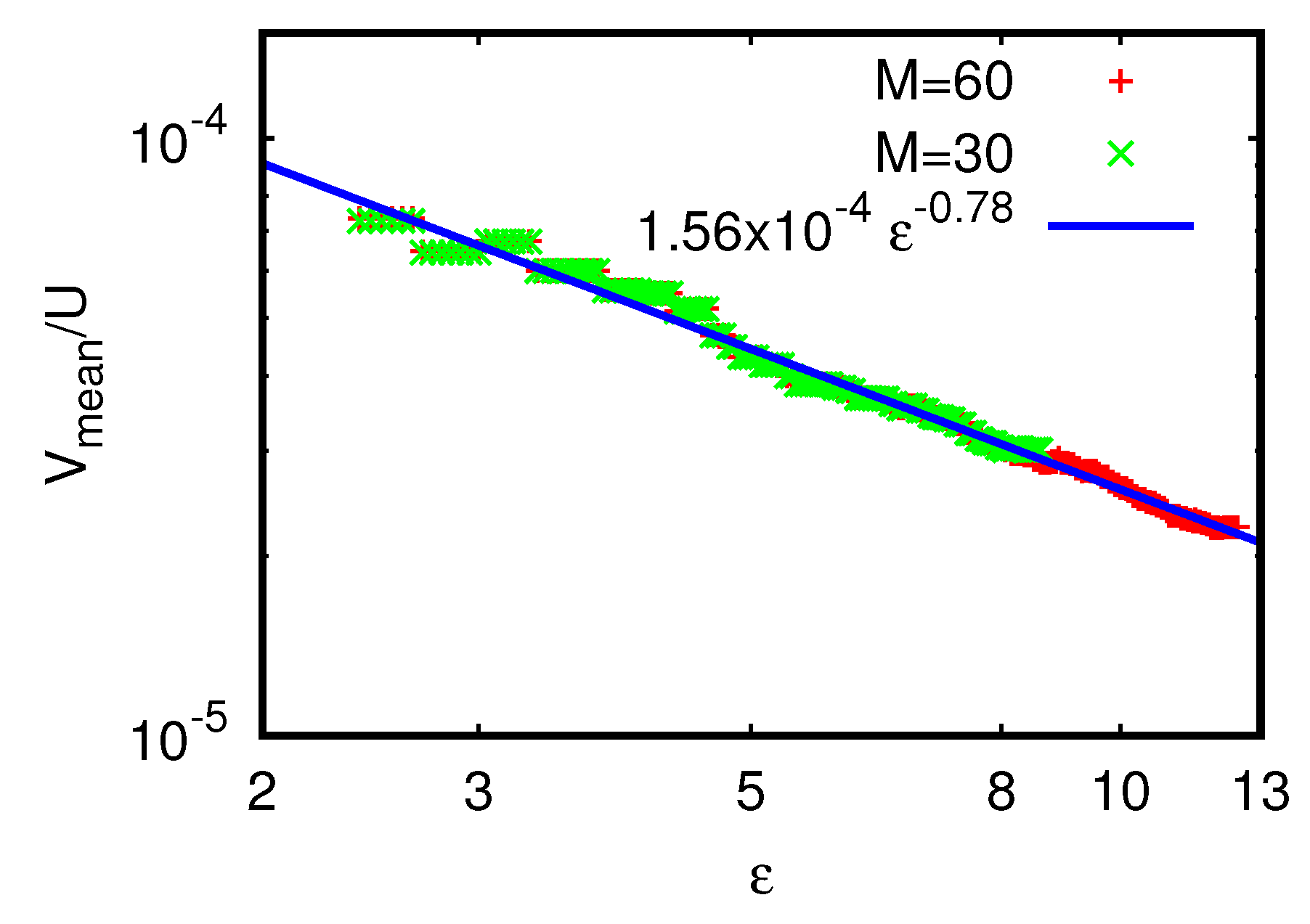
Figure 13 shows the dependence of
on
A for the usual four initial states together with the fit
for the data with initial state
. The values of the parameters
,
for this and the other initial states are given in
Table 2. Here, the initial quadratic dependence
is highly accurate (with no logarithmic correction). Similarly to
, there is only a slight dependence of the values of
and the fit values on the choice of initial state.
Theoretically, we expect according to Fermi’s Golden rule that:
where
according to the discussion below (
10) and
is the effective two-body density of states for states directly coupled by the interaction such that
(see discussion below (
7)). We note that
is the typical interaction matrix element in Fock space which is slightly larger than
(see the theoretical discussion above for the computation of the coefficient
used in (
10) and Appendix A of [
46]). The factor
is due to our particular definition (Again, for practical reasons and similarly to
, we incorporate in the definition of
the time scale
, i.e.,
usual decay rate found in the literature and meaning that
is defined as the ratio of the usual decay rate over the global spectral bandwidth) of decay rates. The expression of
is actually also valid for larger values of
A provided we use the density of states
in the presence of interactions that provides an additional factor
according to (
10). Therefore, at the band center, we have:
which gives, together with (
8):
For
and
, the square root factor is 1.5 and we have to compare
with the values of
in
Table 1, which are somewhat smaller, probably due to a reduction factor for the energy dependent density of states since the energies of the initial states have a certain distance to the band center. Furthermore, according to (
15), we have to compare
with
which is not perfect but gives the correct order of magnitude. For both parameters, the numerical matching is quite satisfactory taking into account the very simple argument using the same typical value of the interaction matrix elements for all cases of initial states.
Finally, we mention that, for the three Åberg parameter values
,
,
used in
Figure 8 and
Figure 9, we have typical decay times in units of
being
, 30, 3, respectively (with some modest fluctuations depending on initial states). These values match quite well the observed time values at which the initially occupied orbitals start to decay (see the above discussion of
Figure 8 and
Figure 9).
3.8. Time Evolution of Density Matrix and Spatial Density
We now turn to the effects of the many-body time evolution in position space (see for example
Figure 1). For this, we compute the spatial density
where
is the one-particle eigenstate of orbital
k, with some examples shown in
Figure 1. Here,
denotes the usual fermion field operators (in the case of continuous
x,
y variables) or standard fermion operators for discrete position basis states (when using a discrete grid for
x and
y positions as we did for the numerical solution of the non-interacting Sinai oscillator model in
Section 2). The sum over orbital index
k in (
16) requires in principle a sum over a
full complete basis set of orbitals with infinite number (case of continuous
x,
y values) or a very large number (case of discrete
x-
y grid) significantly larger than the very modest number of orbitals
M we used for the numerical solution of the many-body Sinai oscillator.
However, we can simply state that, in our model, by construction, all orbitals with
are never occupied such that, in the expectation value for
only the values
are necessary. Taking this into account together with the fact that the one-particle eigenstates are real valued, we obtain the more explicit expression:
where
is the density matrix in orbital representation generalizing the occupation numbers
which are its diagonal elements. Due to the complex phases of
(when expanded in the usual basis of non-interacting many-body states), the density matrix is complex valued but hermitian:
. Therefore, its anti-symmetric imaginary part does not contribute in
. We have numerically evaluated (
17) and we present in
Figure 14 color plots of the density matrix and the spatial density
for
, the initial state
and four time values
. Since the density
does not provide a lot of spatial structure, we also show in
Figure 14 the density difference with respect to the initial condition
which reveals more of its structure (figures and videos for the time evolution of this and other cases are available for download at the web page [
71]).
At the time , density matrix and spatial density are essentially identical to the initial condition at . For , we see a non-trivial structure since there is a small difference with the initial condition and the color plot simply amplifies small maximal amplitudes to maximal color values (red/yellow for strongest positive/negative values even if the latter are small in an absolute scale). The density matrix is diagonal and its diagonal values are either 1 (for initially occupied orbitals) or 0 (for initially empty orbitals) and the spatial density simply gives the sum of densities due to the occupied eigenstates.
At
, we see a non-trivial structure in the density matrix with a lot of non-vanishing values in certain off-diagonal elements. Furthermore, the orbitals 13 and 16 are also slightly excited (see also discussion of
Figure 8) and there is a significant change of the spatial density.
Later, at , the number/values of off-diagonal elements in the density matrix is somewhat reduced, but they are still visible. Especially between orbitals 12 and 13 as well as 14 and 15, there is a rather strong coupling. Orbital 13 is now more strongly excited than the initially excited orbital 12. In addition, orbitals 14 and 15 are quite strong. The spatial density has become smoother and the structure of is roughly close to the case at but with some significant differences.
Finally, at , the density matrix seems be diagonal with values close to the thermalized values. There is a further increase of the density smoothness and has a similar but different structure as for or .
Apparently, at intermediate times
, there are some quantum correlations between certain orbitals, visible as off-diagonal elements in the density matrix which disappear for later times. This kind of decoherence is similar to the exponential decay observed in [
46] for the off-diagonal element of the
density matrix for a qubit coupled to a chaotic quantum dot or the SYK black hole. However, to study this kind of decoherence more carefully in the context here, it would be necessary to use as initial state a non-trivial linear combination of two non-interacting eigenstates and not to rely on the creation of modest off-diagonal elements for intermediate time scales as we see here.
The spatial density is globally rather smooth and typically quite well given by the “classical” relation
in terms of the time dependent occupation numbers. Only for intermediate time scales with more visible quantum coherence (more off-diagonal elements
), this relation is less accurate. However, at
, the density still exhibits small but regular fluctuations in its detail structure as can be seen in the structure of
for later time scales. A closer inspection of the data (for time values not shown in
Figure 14) also shows that, even at long time scales, there are significant fluctuations of
when
t is slightly changed by a few multiples of
.
In
Figure 14, we also show for comparison the theoretical thermalized quantities where, in (
17), the density matrix is replaced by a diagonal matrix with entries being the thermalized occupations numbers
at energy
, which is the typical total one-particle average energy of
for the long time limit
showing that, at
, the state is very close to thermalization but still with small significant differences (see also discussion of
Figure 7 for this case).
We may also generalize the spatial density (
16) to a spatial density correlator which we define as:
depending on initial
and final position
. As an illustration, we choose the fixed value
which is very close to the maximal position (center of the red area) of the one-particle ground state
visible in panel (
a) of
Figure 1. The spatial density correlator is potentially complex with a non-vanishing imaginary part in case of non-vanishing off-diagonal matrix elements of
for
. In
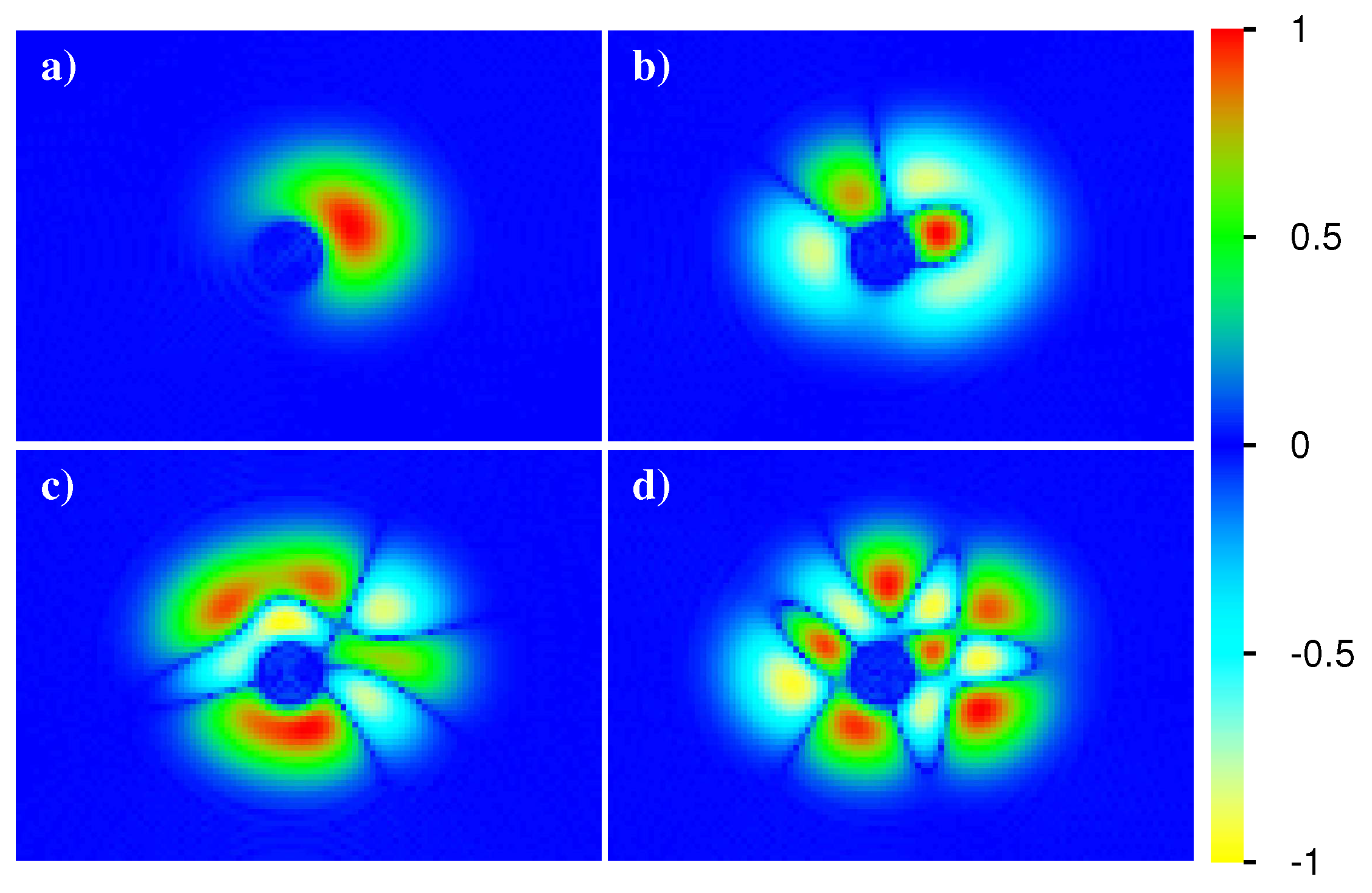
Figure 15, we present density plots of absolute value, real and imaginary part of
in
plane and with the given value
for the same parameters of
Figure 14 (concerning initial state, Åberg parameter, time values and also thermalized case). However, for the thermalized case, the density matrix is diagonal by construction and the imaginary part of
vanishes (giving a blue panel due to zero values).
There are significant time dependent fluctuations of
for all time scales with real part and absolute value being dominated by rather strong maximal values for positions close to the initial position. However, the imaginary part (which vanishes at
and is typically smaller than the values in maximum domain of a real part) shows a more interesting structure since the color plot amplifies small amplitudes (in absolute scale). Apart from this, the absolute and real part values for positions outside the maximum domain (far away from the initial position) seem to decay for long timescales, which is also confirmed by the thermalized case. Even though the case for
seems to be rather close to the thermalized case (for absolute value and real part), there are still differences that are more significant here as in
Figure 14.
Globally, the obtained results show that the dynamical thermalization takes place well, leading to the usual Fermi–Dirac thermal distribution when the Åberg criterion is satisfied and interactions are sufficiently strong to drive the system into the thermal state.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}