
Electron Lone-Pairs Stereochemistry and Drastic van der Waals and Pressure Effects in AsF3 from First Principles


Abstract
:1. Introduction
2. Theoretical and Computational Frameworks
3. Stereochemical Considerations
3.1. Arsenic Trifluoride AsF3E
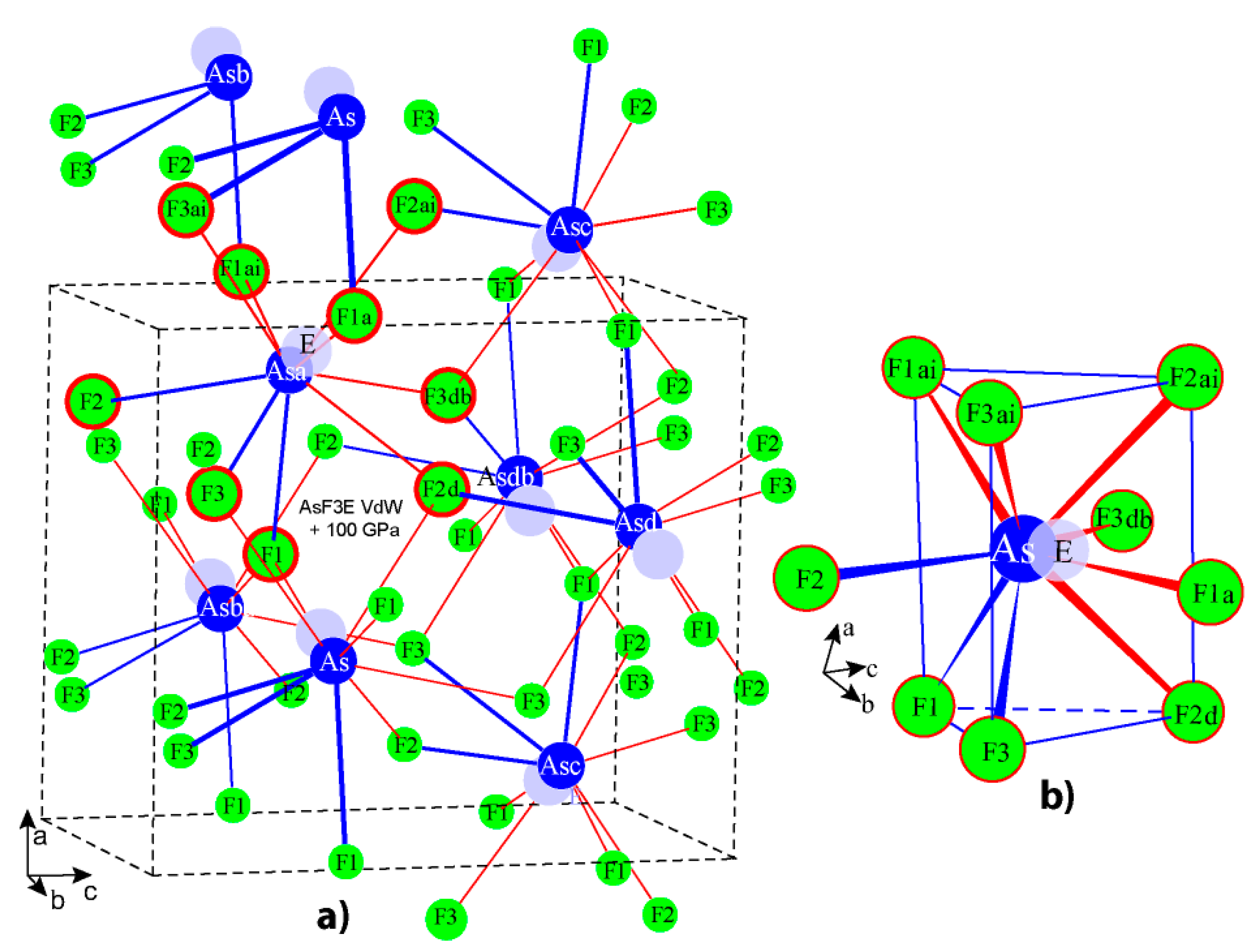
3.2. Crystal Formation and Structure
3.3. Description and Crystal Chemistry Rationale
4. Results for Stereochemical Assessments
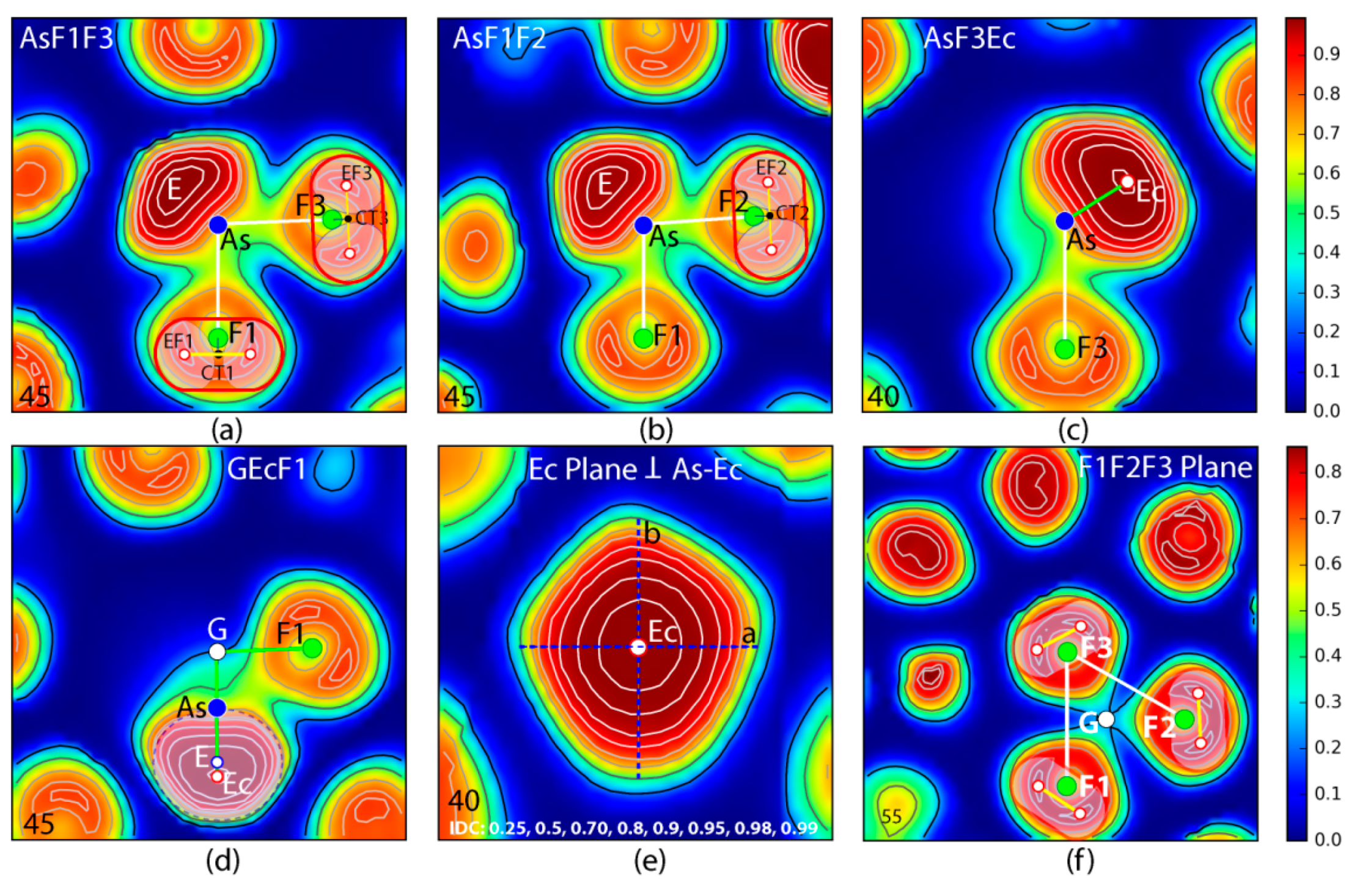
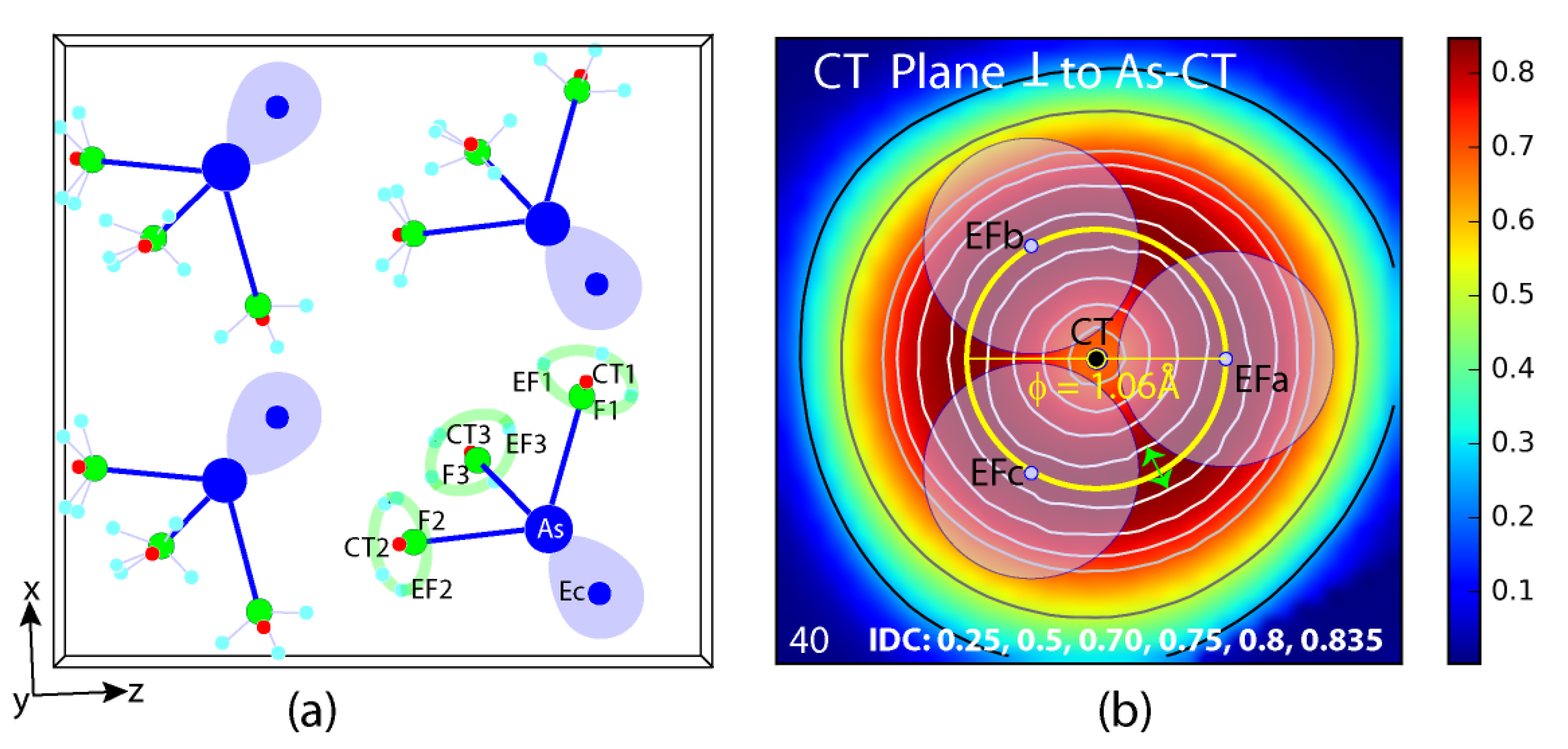
4.1. Electron Localization Function ELF
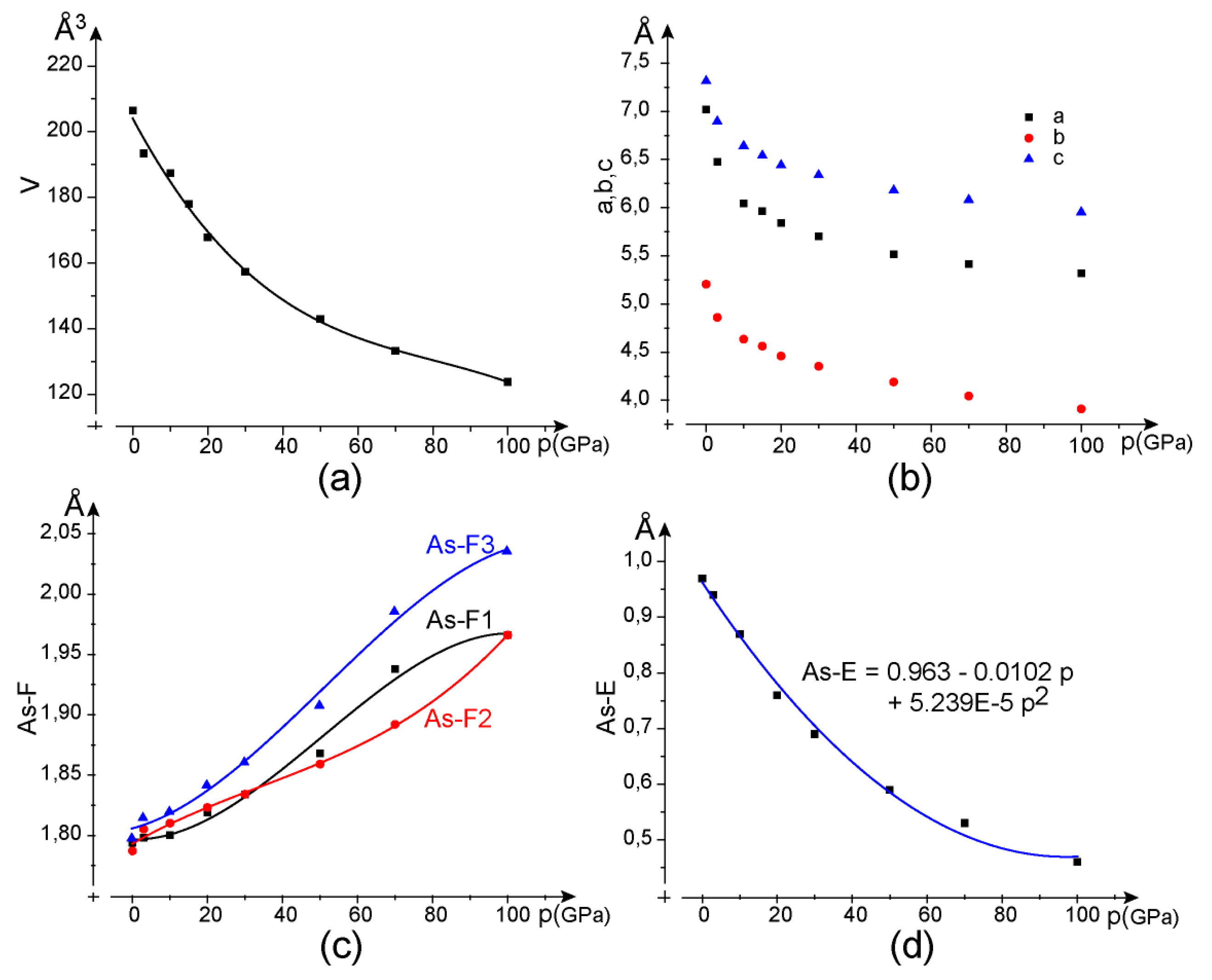
4.2. Calculations Accounting for van der Waals Forces and Simulation of Various Pressures
5. Electronic Band Structures, Density of States and Chemical Bonding
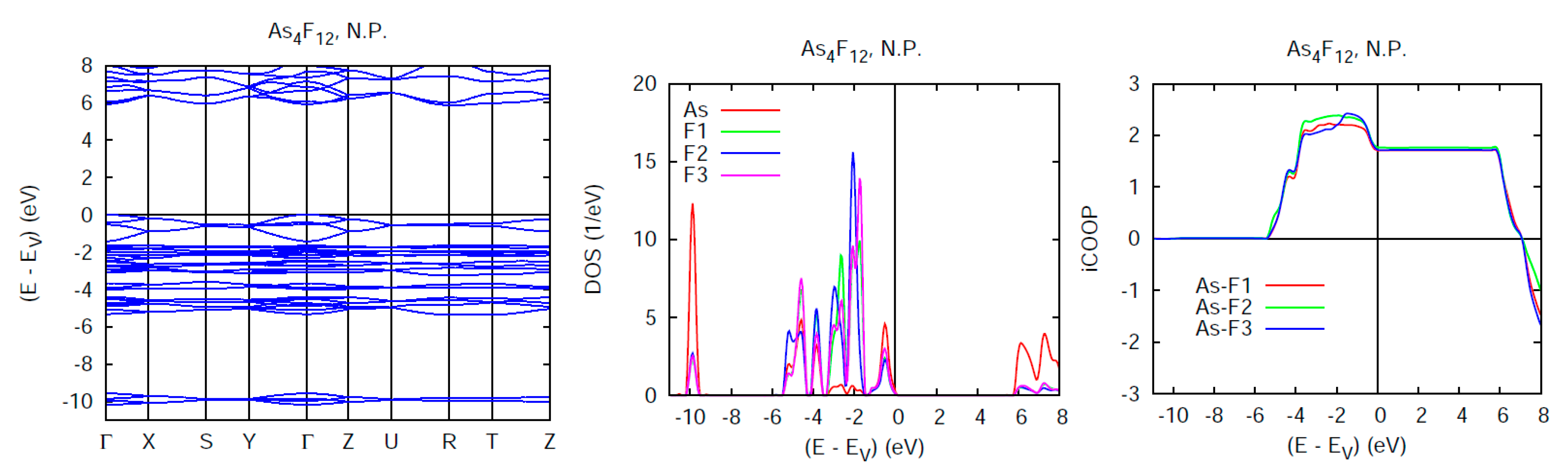
5.1. Normal Pressure Results
5.2. Pressure Effects
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galy, J.; Matar, S.F. ns2np4 (n = 4, 5) lone pair triplets whirling in M*F2E3 (M* = Kr, Xe): Stereochemistry and ab initio analyses. Solid State Sci. 2017, 64, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Galy, J.; Matar, S.F. Joint stereochemical and ab initio overview of SnII electron lone pairs (E) and F−(E) triplets effects on the crystal networks, the bonding and the electronic structures in a family of tin fluorides. J. Prog. Solid State Chem. 2019, 56, 100252. [Google Scholar] [CrossRef]
- Clippard, F.B., Jr.; Bartell, L.S. Molecular structures of arsenic trifluoride and arsenic pentafluoride as determined by elec-tron diffraction. Inorg. Chem. 1970, 9, 805–811. [Google Scholar] [CrossRef]
- Grimme, S. Semi-empirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Greis, O.; Martinez Ripoll, M. Crystal structure of BiF3. Z. Anorg. Allg. Chem. 1977, 436, 105–112. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. The Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total-energy calculations for metals and semiconductors using a plane-wave basis set. Phys. Rev. B Condens. Matter Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, J. From ultrasoft pseudopotentials to the projector augmented wave. Phys. Rev. B Condens. Matter Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. The projected augmented wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MEDEA simulation Package. Materials Design Co. Available online: https://www.materialsdesign.com/software-releases (accessed on 7 August 2021).
- Bezugly, B.V.; Kohout, P.; Wagner, F.R. Electron localizability indicators ELI-D and ELIA for highly correlated wavefunctions of homonuclear dimers. II. N2, O2, F2, and Ne2. J. Comput. Chem. 2010, 31, 1404–1519. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397. [Google Scholar] [CrossRef]
- Eyert, V. Basic notions and applications of the augmented spherical wave method. Int. J. Quant. Chem. 2000, 77, 1007–1012. [Google Scholar] [CrossRef]
- Hoffmann, R. How chemistry and physics meet in the solid state. Angew. Chem. Int. Ed. Engl. 1987, 26, 846–848. [Google Scholar] [CrossRef]
- Pauling, L.; Brockway, L.O. The radial distribution method of interpretation of electron diffraction photographs of gas molecules. J. Am. Chem. Soc. 1935, 57, 2684. [Google Scholar] [CrossRef]
- Enjalbert, R.; Galy, J. Structure cristalline du trifluorure d’arsenic AsF3 à 193K. C.R. Acad. Sc. 1979, C289, 441–443. [Google Scholar]
- Galy, J.; Enjalbert, R. Crystal chemistry of the Va element trihalides: Lone pair, stereochemistry, and structural relationships. J. Solid State Chem. 1982, 44, 1–23. [Google Scholar] [CrossRef]
- Hund Von, F.; Fricke, R. Der Kristallbau von α-BiF3. Z. Anorg. Allg. Chem. 1949, 258, 198–204. [Google Scholar] [CrossRef]
- Croatto, U.N. The gananite mineral. Gazs. Chim. Ital. 1944, 71, 20. [Google Scholar]
- Hyde, B.G.; Andersson, S. Inorganic Crystal Structures; Wiley-Interscience Publication: Hoboken, NJ, USA, 1989; pp. 122–123. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AsF3E/Cell (RX [21]) | a (Å) | b (Å) | c (Å) | V (Å3) | Z | rV (F,E) (Å3) |
|---|---|---|---|---|---|---|
| RX [21] AsF3E | 7.018 | 5.205 | 7.315 | 267.2 | 4 | 16.7 |
| Atom coordinates of the molecule and E localization (Figure 1) | Atoms | x | y | z | Wyckoff | |
| As | 0.7862 | 0.5338 | 0.25 | 4a | ||
| F1 | 0.5736 | 0.3995 | 0.3065 | 4a | ||
| F2 | 0.8077 | 0.3740 | 0.0490 | 4a | ||
| F3 | 0.6792 | 0.7956 | 0.1493 | 4a | ||
| E | 0.8885 | 0.5529 | 0.3371 | 4a | ||
| AsF3E/Cells | a (Å) | b (Å) | c (Å) | V (Å3) | Z | rV (F,E) (Å3) |
| AsF3E (DE) | 6.632 | 4.957 | 7.013 | 230.5 | 4 | 14.4 |
| AsF3E (DEW) | 6.346 | 4.780 | 6.806 | 206.4 | 4 | 12.9 |
| DEW+ 3 GPa | 6.174 | 4.681 | 6.691 | 193.4 | 4 | 12.1 |
| DEW+ 30 GPa | 5.701 | 4.353 | 6.339 | 157.3 | 4 | 9.8 |
| DEW+ 100 GPa | 5.319 | 3.910 | 5.954 | 123.8 | 4 | 7.7 |
| Data (Å, °, GPa) | RX [21] (Å, °) | DEW + GPa (Å, °, 3 GPa) | DEW + GPa (Å, °, 30 GPa) | DEW + GPa (Å, °, 100 GPa) | ||
| As-Ec | 0.97 | 0.94 | 0.69 | 0.46 | ||
| As-F1 | 1.699 | 1.798 | 1.834 | 1.966 | ||
| As-F2 | 1.696 | 1.805 | 1.834 | 1.966 | ||
| As-F3 | 1.722 | 1.815 | 1.861 | 2.036 | ||
| F1-F2 | 2.503 | 2.593 | 2.535 | 2.380 | ||
| F2-F3 | 2.483 | 2.501 | 2.395 | 2.174 | ||
| F3-F1 | 2.474 | 2.543 | 2.491 | 2.381 | ||
| Asb-F1a | 3.054 | 2.546 | 2.253 | 1.918 | ||
| As-F2c | 2.890 | 2.422 | 2.218 | 2.032 | ||
| F-As-F angles (°) | 95.0, 93.2, 92.7 | 89.5, 87.4, 92.1 | 87.4, 80.8, 84.8 | 74.5, 65.8, 73.0 | ||
| As-Ec | 0.97 | Ec-F1,2,3 | 2.38 | ∠FAsF * | 93.6 * |
| F-{EF} (1,2,3) | 0.57 | F -CT (1,2,3) | 0.20 | Ec a/b/c ellipsoid | 0.94/1.03/0.85 |
| CT-{EF}(1,2,3) | 0.53 | As-CT (1,2,3) | 1.91 | {EF}a/b/c ellipsoid | 0.41/0.41/0.55 |
| T. Rd. rect. {EF} (Figure 2a,b) L/H/R | 1.98/1.10/0.75 | ϕT.ext.{EF3} blue dotted circle | 1.98 | T. gyr. ϕ yellow stick or ϕ circle | 1.06 |
| rEc | 0.94 | r{EF} | 0.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galy, J.; Matar, S.F. Electron Lone-Pairs Stereochemistry and Drastic van der Waals and Pressure Effects in AsF3 from First Principles. Condens. Matter 2021, 6, 31. https://doi.org/10.3390/condmat6030031
Galy J, Matar SF. Electron Lone-Pairs Stereochemistry and Drastic van der Waals and Pressure Effects in AsF3 from First Principles. Condensed Matter. 2021; 6(3):31. https://doi.org/10.3390/condmat6030031
Chicago/Turabian StyleGaly, Jean, and Samir F. Matar. 2021. "Electron Lone-Pairs Stereochemistry and Drastic van der Waals and Pressure Effects in AsF3 from First Principles" Condensed Matter 6, no. 3: 31. https://doi.org/10.3390/condmat6030031
APA StyleGaly, J., & Matar, S. F. (2021). Electron Lone-Pairs Stereochemistry and Drastic van der Waals and Pressure Effects in AsF3 from First Principles. Condensed Matter, 6(3), 31. https://doi.org/10.3390/condmat6030031