
Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics


 ,
, 
Abstract
:1. Introduction
2. Process of Apoptosis
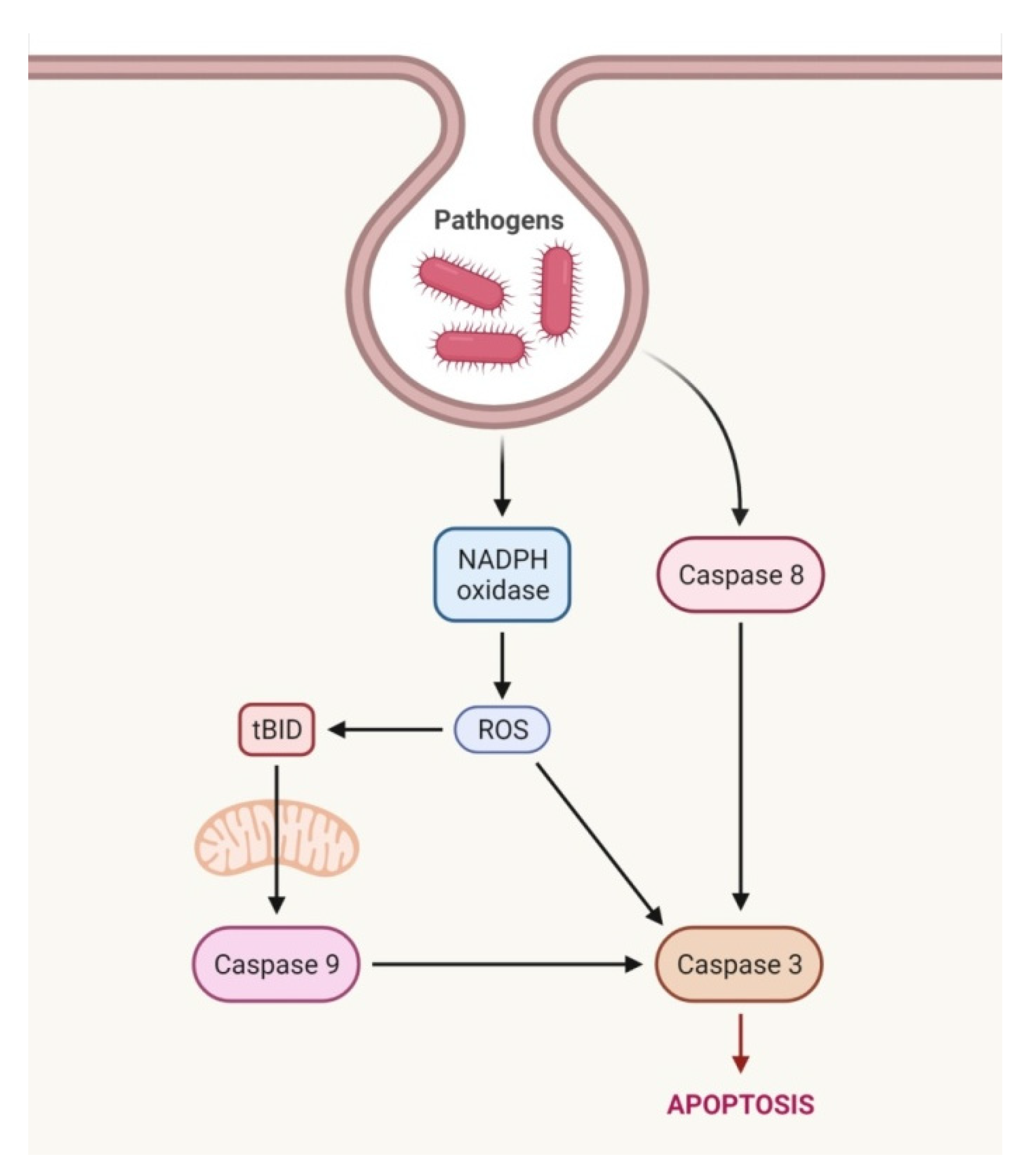
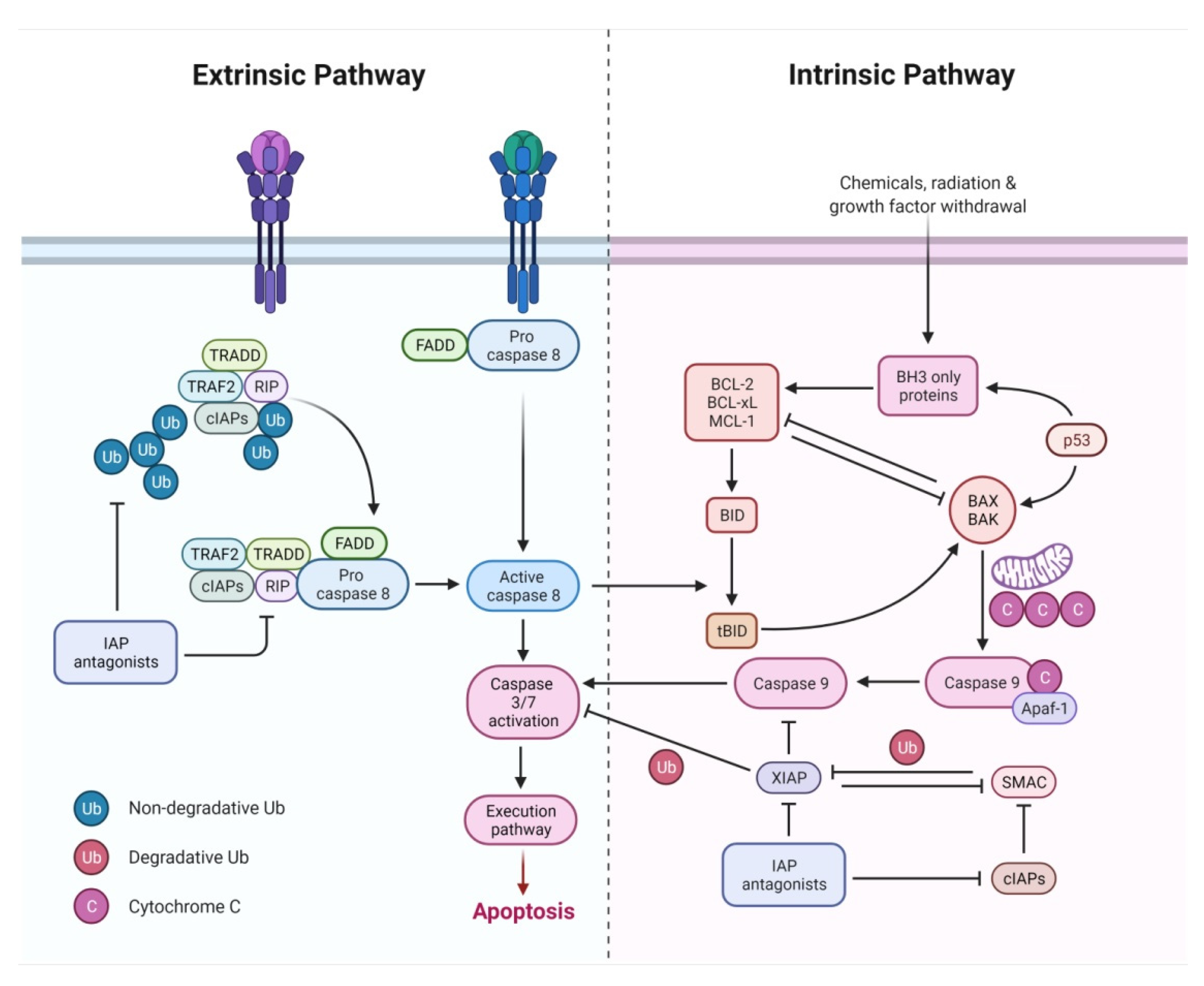
2.1. Extrinsic Pathway
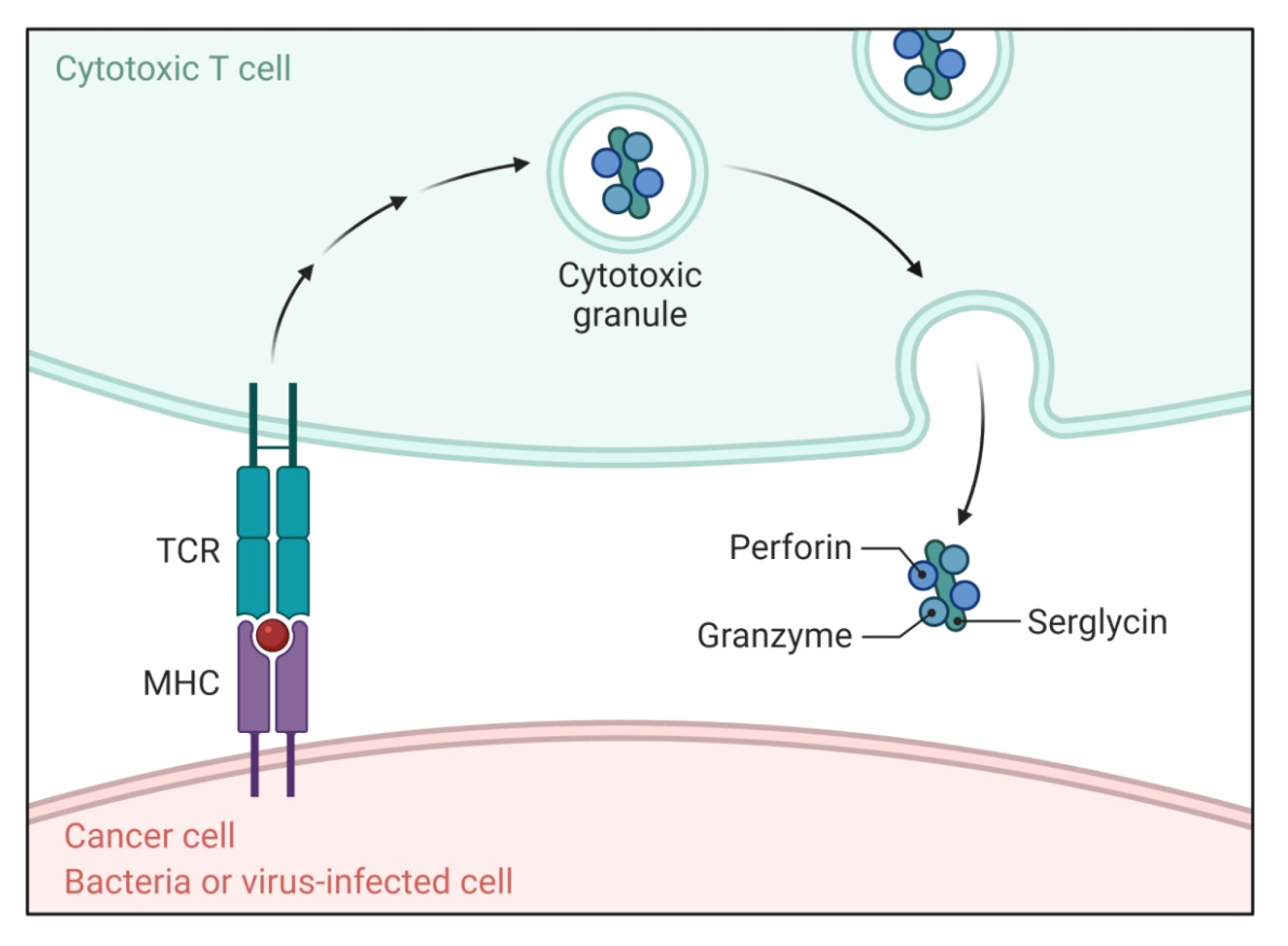
2.2. Perforin/Granzyme Pathway
2.3. Intrinsic Pathway
2.4. Execution Pathway
3. Caspases
4. Apoptosis-Associated Protein Domains
4.1. Death Domain Proteins
4.2. Death Effector Domain Proteins
4.3. CARD-Family Proteins
4.4. Inhibitor of Apoptosis Proteins
4.5. BCL-2 Family Proteins
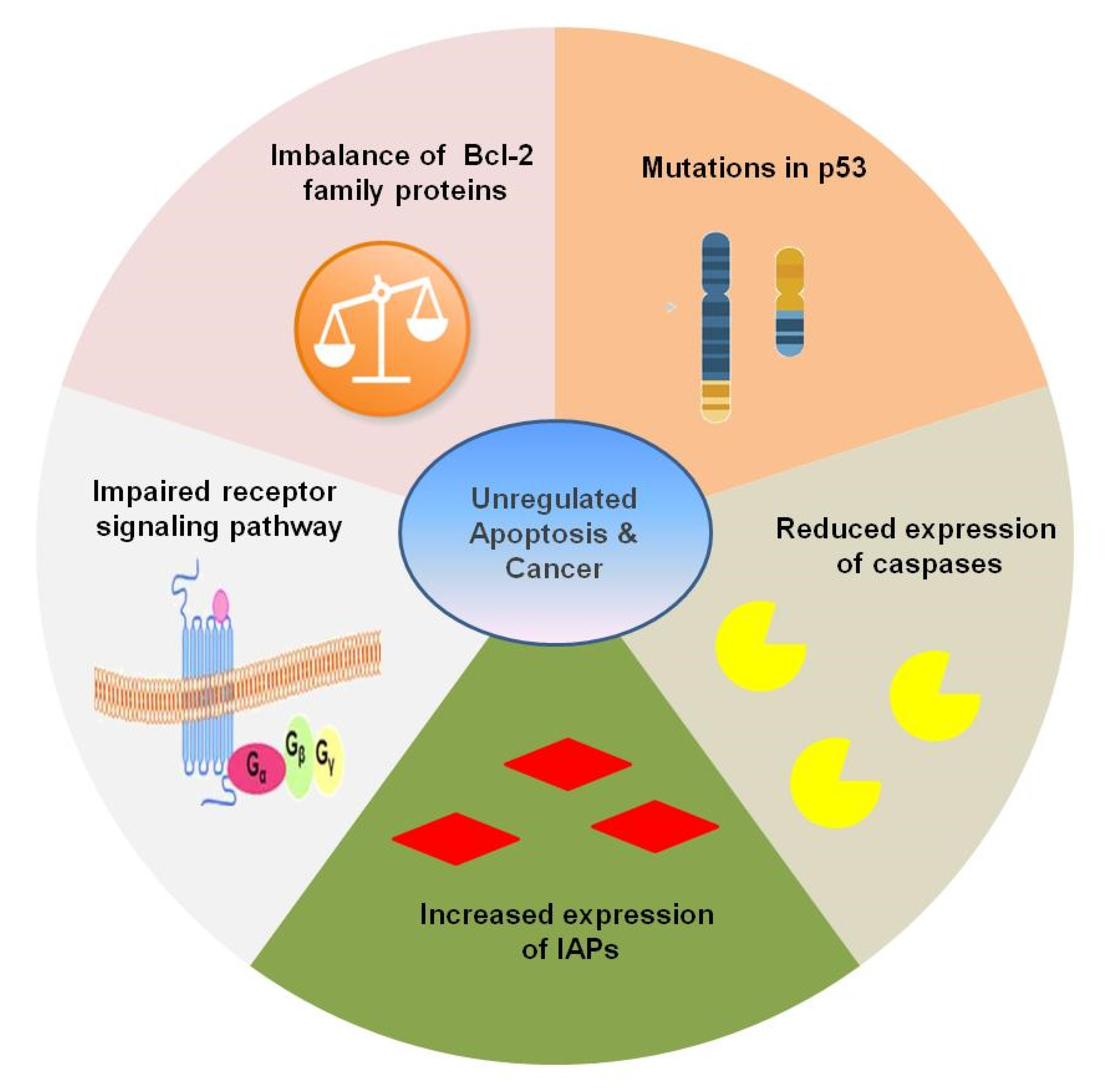
5. Apoptosis and Carcinogenesis
6. Targeting Apoptosis
6.1. Approaches Targeting Intrinsic Pathway of Apoptosis
6.1.1. BH3 Mimetics
6.1.2. MCL-1 Inhibitors
6.1.3. IAP Inhibitors
6.2. Approaches Targeting Extrinsic Pathway
6.2.1. Death Receptor Agonists
6.2.2. Tumor Suppressor Pathways
6.2.3. Epigenetic Approaches Targeting Apoptosis
6.2.4. Chaperons Targeting Apoptosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Paweletz, N. Walther Flemming: Pioneer of mitosis research. Nat. Rev. Mol. Cell Biol. 2001, 2, 72–75. [Google Scholar] [CrossRef]
- Kerr, J.F.R. History of the events leading to the formulation of the apoptosis concept. Toxicology 2002, 181, 471–474. [Google Scholar] [CrossRef]
- Horvitz, H.R. The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 377–385. [Google Scholar] [CrossRef]
- Formigli, L.; Papucci, L.; Tani, A.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Zecchi Orlandini, S. Aponecrosis: Morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell. Physiol. 2000, 182, 41–49. [Google Scholar] [CrossRef]
- Singh, V.; Khurana, A.; Allawadhi, P.; Banothu, A.K.; Bharani, K.K.; Weiskirchen, R. Emerging role of PD-1/PD-L1 inhibitors in chronic liver diseases. Front. Pharmacol. 2021, 12, 790963. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Debnath, J.; Baehrecke, E.H.; Kroemer, G. Does autophagy contribute to cell death? Autophagy 2005, 1, 66–74. [Google Scholar] [CrossRef]
- Norbury, C.J.; Hickson, I.D. Cellular responses to DNA damage. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 367–401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Wu, Y.; Lai, H.W.; Yew, D.T. Apoptosis–a brief review. Neuroembryol. Aging 2004, 3, 47–59. [Google Scholar] [CrossRef]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 238–259. [Google Scholar] [CrossRef] [Green Version]
- Allawadhi, P.; Khurana, A.; Allwadhi, S.; Joshi, K.; Packirisamy, G.; Bharani, K.K. Nanoceria as a possible agent for the management of COVID-19. Nano Today 2020, 35, 100982. [Google Scholar] [CrossRef]
- Allawadhi, P.; Singh, V.; Khurana, I.; Rawat, P.S.; Renushe, A.P.; Khurana, A.; Navik, U.; Allwadhi, S.; Karlapudi, S.K.; Banothu, A.K.; et al. Decorin as a possible strategy for the amelioration of COVID-19. Med. Hypotheses 2021, 152, 110612. [Google Scholar] [CrossRef]
- Khurana, I.; Allawadhi, P.; Khurana, A.; Srivastava, A.K.; Navik, U.; Banothu, A.K.; Bharani, K.K. Can bilirubin nanomedicine become a hope for the management of COVID-19? Med. Hypotheses 2021, 149, 110534. [Google Scholar] [CrossRef]
- Khurana, A.; Sayed, N.; Allawadhi, P.; Weiskirchen, R. It’s all about the spaces between cells: Role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 2021, 9, 728. [Google Scholar] [CrossRef]
- BioRender. Create Professional Science Figures in Minutes. Available online: https://biorender.com/ (accessed on 11 January 2022).
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Navik, U.; Sheth, V.G.; Khurana, A.; Jawalekar, S.S.; Allawadhi, P.; Gaddam, R.R.; Bhatti, J.S.; Tikoo, K. Methionine as a double-edged sword in health and disease: Current perspective and future challenges. Ageing Res. Rev. 2021, 72, 101500. [Google Scholar] [CrossRef]
- Allawadhi, P.; Khurana, A.; Allwadhi, S.; Navik, U.S.; Joshi, K.; Banothu, A.K.; Bharani, K.K. Potential of electric stimulation for the management of COVID-19. Med. Hypotheses 2020, 144, 110259. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Smith, L.K.; Cidlowski, J.A. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog. Brain Res. 2010, 182, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Häcker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef]
- Papaliagkas, V.; Anogianaki, A.; Anogianakis, G.; Ilonidis, G. The proteins and the mechanisms of apoptosis: A mini-review of the fundamentals. Hippokratia 2007, 11, 108–113. [Google Scholar]
- Hongmei, Z. Extrinsic and Intrinsic Apoptosis Signal Pathway Review. In Apoptosis and Medicine; Ntuli, T., Ed.; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Allawadhi, P.; Khurana, A.; Banothu, A.K.; Bharani, K.K. Critical neurological features of COVID-19: Role of imaging methods and biosensors for effective diagnosis. Sens. Int. 2021, 2, 100098. [Google Scholar] [CrossRef]
- Wyllie, A.H. “Where, O death, is thy sting?” A brief review of apoptosis biology. Mol. Neurobiol. 2010, 42, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Allawadhi, P.; Singh, V.; Govindaraj, K.; Khurana, I.; Sarode, L.P.; Navik, U.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K.; Khurana, A. Biomedical applications of polysaccharide nanoparticles for chronic inflammatory disorders: Focus on rheumatoid arthritis, diabetes and organ fibrosis. Carbohydr. Polym. 2021, 281, 118923. [Google Scholar] [CrossRef]
- Allawadhi, P.; Khurana, A.; Sayed, N.; Godugu, C.; Vohora, D. Ameliorative effect of cerium oxide nanoparticles against Freund’s complete adjuvant-induced arthritis. Nanomedicine 2022, 17, 383–404. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Martinvalet, D.; Zhu, P.; Lieberman, J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 2005, 22, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and-independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008, 19, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Smyth, M. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.H.; Ley, T.J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef]
- Brunner, T.; Wasem, C.; Torgler, R.; Cima, I.; Jakob, S.; Corazza, N. Fas (CD95/Apo-1) Ligand regulation in T cell homeostasis, cell-mediated cytotoxicity and immune pathology. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2003; Volume 15, pp. 167–176. [Google Scholar] [CrossRef]
- Goping, I.S.; Barry, M.; Liston, P.; Sawchuk, T.; Constantinescu, G.; Michalak, K.M.; Shostak, I.; Roberts, D.; Hunter, A.M.; Korneluk, R.; et al. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity 2003, 18, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.J.; Sasaki, T.; Elia, A.J.; Cheng, H.-Y.M.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial protein that promotes cytochrome c–dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, G.; Van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, A.M. The Apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-T.; Newland, A.C.; Jia, L. Bax conformational change is a crucial step for PUMA-mediated apoptosis in human leukemia. Biochem. Biophys. Res. Commun. 2003, 310, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [Green Version]
- Sakahira, H.; Enari, M.; Nagata, S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998, 391, 96–99. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [Green Version]
- Salvesen, G.S. Caspases and apoptosis. Essays Biochem. 2002, 38, 9–19. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis 2002, 7, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Grütter, M.G. Caspases: Key players in programmed cell death. Curr. Opin. Struct. Biol. 2000, 10, 649–655. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Salvesen, G.S. Properties of the caspases. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1998, 1387, 17–31. [Google Scholar] [CrossRef]
- Molla, M.D.; Akalu, Y.; Geto, Z.; Dagnew, B.; Ayelign, B.; Shibabaw, T. Role of caspase-1 in the pathogenesis of inflammatory-associated chronic noncommunicable diseases. J. Inflamm. Res. 2020, 13, 749–764. [Google Scholar] [CrossRef]
- Mancini, M.; Machamer, C.E.; Roy, S.; Nicholson, D.W.; Thornberry, N.A.; Casciola-Rosen, L.A.; Rosen, A. Caspase-2 Is Localized at the Golgi Complex and Cleaves Golgin-160 during Apoptosis. J. Cell Biol. 2000, 149, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, function, and biotechnological aspects. Biotechnol. Appl. Biochem. 2021; Online ahead of print. [Google Scholar] [CrossRef]
- Chen, Q.; Shi, P.; Wang, Y.; Zou, D.; Wu, X.; Wang, D.; Hu, Q.; Zou, Y.; Huang, Z.; Ren, J.; et al. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J. Mol. Cell Biol. 2018, 11, 496–508. [Google Scholar] [CrossRef]
- Aslan, A.; Gok, O.; Erman, O.; Kuloglu, T. Ellagic acid impedes carbontetrachloride-induced liver damage in rats through suppression of NF-κB, Bcl-2 and regulating Nrf-2 and caspase pathway. Biomed. Pharmacother. 2018, 105, 662–669. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.I.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK–caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Liao, B.; Sun, Q.; Yuan, Y.; Yin, Y.; Qiao, J.; Jiang, P. Histone deacetylase inhibitor MGCD0103 causes cell cycle arrest, apoptosis, and autophagy in liver cancer cells. J. Cancer 2020, 11, 1915–1926. [Google Scholar] [CrossRef]
- Mandal, P.; Feng, Y.; Lyons, J.D.; Berger, S.B.; Otani, S.; DeLaney, A.; Tharp, G.K.; Maner-Smith, K.; Burd, E.M.; Schaeffer, M.; et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity 2018, 49, 42–55.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Gu, X.; Fang, J.; Cai, D.; Zuo, Z.; Liang, S.; Cui, H.; Deng, J.; Ma, X.; Geng, Y.; et al. Effect of intranasal instillation of Escherichia coli on apoptosis of spleen cells in diet-induced-obese mice. Sci. Rep. 2020, 10, 5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meynier, S.; Rieux-Laucat, F. FAS and RAS related Apoptosis defects: From autoimmunity to leukemia. Immunol. Rev. 2018, 287, 50–61. [Google Scholar] [CrossRef]
- Yi, Y.-S. Caspase-11 Noncanonical inflammasome: A novel key player in murine models of neuroinflammation and multiple sclerosis. Neuroimmunomodulation 2021, 28, 195–203. [Google Scholar] [CrossRef]
- Song, J.; Zhang, Q.; Wang, S.; Yang, F.; Chen, Z.; Dong, Q.; Ji, Q.; Yuan, X.; Ren, D. Cleavage of caspase-12 at Asp94, mediated by endoplasmic reticulum stress (ERS), contributes to stretch-induced apoptosis of myoblasts. J. Cell. Physiol. 2018, 233, 9473–9487. [Google Scholar] [CrossRef]
- Markiewicz, A.; Sigorski, D.; Markiewicz, M.; Owczarczyk-Saczonek, A.; Placek, W. Caspase-14—From biomolecular basics to clinical approach. A review of available data. Int. J. Mol. Sci. 2021, 22, 5575. [Google Scholar] [CrossRef]
- Kitson, J.; Raven, T.; Jiang, Y.-P.; Goeddel, D.V.; Giles, K.M.; Pun, T.; Grinham, C.J.; Brown, R.; Farrow, S.N. A death-domain-containing receptor that mediates apoptosis. Nature 1996, 384, 372–375. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.J.; Matsuo, H.; Duan, H.; Wagner, G. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 1998, 94, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvez, F.; Ashkenazi, A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010, 29, 4752–4765. [Google Scholar] [CrossRef] [Green Version]
- Park, H.H.; Lo, Y.-C.; Lin, S.-C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, A.; Allawadhi, P.; Khurana, I.; Allwadhi, S.; Weiskirchen, R.; Banothu, A.K.; Chhabra, D.; Joshi, K.; Bharani, K.K. Role of nanotechnology behind the success of mRNA vaccines for COVID-19. Nano Today 2021, 38, 101142. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Ferguson, T.A. The role of FasL-induced apoptosis in immune privilege. Immunol. Today 1997, 18, 240–244. [Google Scholar] [CrossRef]
- Valmiki, M.G.; Ramos, J.W. Death effector domain-containing proteins. Cell. Mol. Life Sci. 2008, 66, 814–830. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, B.C.; Lee, J.C.; Alappat, E.C.; Peter, M.E. The death effector domain protein family. Oncogene 2003, 22, 8634–8644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibbetts, M.D.; Zheng, L.; Lenardo, M.J. The death effector domain protein family: Regulators of cellular homeostasis. Nat. Immunol. 2003, 4, 404–409. [Google Scholar] [CrossRef]
- Aravind, L.; Dixit, V.M.; Koonin, E.V. The domains of death: Evolution of the apoptosis machinery. Trends Biochem. Sci. 1999, 24, 47–53. [Google Scholar] [CrossRef]
- Hofmann, K.; Bucher, P.; Tschopp, J. The CARD domain: A new apoptotic signalling motif. Trends Biochem. Sci. 1997, 22, 155–156. [Google Scholar] [CrossRef]
- Yang, X.; Chang, H.Y.; Baltimore, D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science 1998, 281, 1355–1357. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins—Suppressors of apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998, 273, 7787–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C. The Survivin saga goes in vivo. J. Clin. Investig. 2001, 108, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Kasof, G.M.; Gomes, B.C. Livin, a novel inhibitor of apoptosis protein family member. J. Biol. Chem. 2001, 276, 3238–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucic, D.; Stennicke, H.R.; Pisabarro, M.T.; Salvesen, G.S.; Dixit, V.M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 2000, 10, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Reed, J.C. Bcl-2 family proteins. Oncogene 1998, 17, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [Green Version]
- Burlacu, A. Regulation of apoptosis by Bcl-2 family proteins. J. Cell. Mol. Med. 2003, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Zha, H.; Aime-Sempe, C.; Takayama, S.; Wang, H.G. Structure—function analysis of Bcl-2 family proteins. Mech. Lymph. Act. Immune Regul. 1996, 6, 99–112. [Google Scholar] [CrossRef]
- Karkale, S.; Khurana, A.; Saifi, M.A.; Godugu, C.; Talla, V. Oropharyngeal administration of silica in Swiss mice: A robust and reproducible model of occupational pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 51, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Putcha, G.V.; Le, S.; Frank, S.; Besirli, C.; Clark, K.; Chu, B.; Alix, S.; Youle, R.J.; LaMarche, A.; Maroney, A.C.; et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 2003, 38, 899–914. [Google Scholar] [CrossRef] [Green Version]
- Moldoveanu, T.; Grace, C.R.; Llambi, F.; Nourse, A.; Fitzgerald, P.; Gehring, K.; Kriwacki, R.W.; Green, D.R. BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 2013, 20, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Degli Esposti, M. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Bcl-2-regulated apoptosis: Mechanism and therapeutic potential. Curr. Opin. Immunol. 2007, 19, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011, 278, 403–413. [Google Scholar] [CrossRef]
- Stewart, M.L.; Fire, E.; Keating, A.E.; Walensky, L.D. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 2010, 6, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci. 2009, 66, 1326–1336. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottina, E.; Tischner, D.; Herold, M.J.; Villunger, A. A1/Bfl-1 in leukocyte development and cell death. Exp. Cell Res. 2012, 318, 1291–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Guttridge, D.C.; Mayo, M.W.; Baldwin, A.S., Jr. NF-κB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell. Biol. 1999, 19, 5923–5929. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, L.; Strasser, A.; O’Reilly, L.A.; Hausmann, G.; Adams, J.; Cory, S.; Huang, D. Bim: A novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998, 17, 384–395. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Howells, C.C.; Baumann, W.T.; Samuels, D.C.; Finkielstein, C.V. The Bcl-2-associated death promoter (BAD) lowers the threshold at which the Bcl-2-interacting domain death agonist (BID) triggers mitochondria disintegration. J. Theor. Biol. 2011, 271, 114–123. [Google Scholar] [CrossRef]
- Coultas, L.; Bouillet, P.; Stanley, E.G.; Brodnicki, T.C.; Adams, J.; Strasser, A. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol. Cell. Biol. 2004, 24, 1570–1581. [Google Scholar] [CrossRef] [Green Version]
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the tip of the balance between life and death. Oncogene 2008, 27, S84–S92. [Google Scholar] [CrossRef]
- Puthalakath, H.; Villunger, A.; O’Reilly, L.A.; Beaumont, J.G.; Coultas, L.; Cheney, R.E.; Huang, D.C.S.; Strasser, A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by Anoikis. Science 2001, 293, 1829–1832. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, A.H. Apoptosis and carcinogenesis. Eur. J. Cell Biol. 1997, 73, 189–197. [Google Scholar] [PubMed]
- Wyllie, A.H.; Bellamy, C.O.; Bubb, V.J.; Clarke, A.R.; Corbet, S.; Curtis, L.; Harrison, D.J.; Hooper, M.L.; Toft, N.; Webb, S.; et al. Apoptosis and carcinogenesis. Br. J. Cancer 1999, 80, 189–197. [Google Scholar]
- Rehman, M.U.; Khan, A.; Imtiyaz, Z.; Ali, S.; Makeen, H.A.; Rashid, S.; Arafah, A. Current Nano-therapeutic Approaches Ameliorating Inflammation in Cancer Progression. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022. [Google Scholar] [CrossRef]
- Shoaib, A.; Tabish, M.; Ali, S.; Arafah, A.; Wahab, S.; Almarshad, F.M.; Rashid, S.; Rehman, M.U. Dietary Phytochemicals in Cancer Signalling Pathways: Role of miRNA Targeting. Curr. Med. Chem. 2021, 28, 8036–8067. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Wani, J.; Majid, S.; Khan, A.; Arafah, A.; Ahmad, A.; Jan, B.; Shah, N.; Kazi, M.; Rehman, M. Clinico-Pathological Importance of miR-146a in Lung Cancer. Diagnostics 2021, 11, 274. [Google Scholar] [CrossRef]
- Rai, K.R.; Moore, J.; Wu, J.; Novick, S.C.; O’Brien, S.M. Effect of the addition of oblimersen (Bcl-2 antisense) to fludarabine/cyclophosphamide for relapsed/refractory chronic lymphocytic leukemia (CLL) on survival in patients who achieve CR/nPR: Five-year follow-up from a randomized phase III study. J. Clin. Oncol. 2008, 26, 7008. [Google Scholar] [CrossRef]
- Abou-Nassar, K.; Brown, J.R. Novel agents for the treatment of chronic lymphocytic leukemia. Clin. Adv. Hematol. Oncol. HO 2010, 8, 886–895. [Google Scholar]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Albershardt, T.C.; Salerni, B.L.; Soderquist, R.S.; Bates, D.J.; Pletnev, A.A.; Kisselev, A.F.; Eastman, A. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J. Biol. Chem. 2011, 286, 24882–24895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocker, M.; Neureiter, D.; Lueders, M.; Zopf, S.; Ganslmayer, M.; Hahn, E.G.; Herold, C.; Schuppan, D. Variants of bcl-2 specific siRNA for silencing antiapoptotic bcl-2 in pancreatic cancer. Gut 2005, 54, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Sengupta, J.; Bu, Y.; Yi, F.; Wang, C.; Shi, Y.; Zhu, Y.; Jiao, Q.; Song, F. Silencing of Bmi-1 gene by RNA interference enhances sensitivity to doxorubicin in breast cancer cells. Indian J. Exp. Biol. 2011, 49. [Google Scholar]
- Roth, J.A.; Nguyen, D.; Lawrence, D.D.; Kemp, B.L.; Carrasco, C.H.; Ferson, D.Z.; Hong, W.K.; Komaki, R.; Lee, J.J.; Nesbitt, J.C.; et al. Retrovirus–mediated wild–type P53 gene transfer to tumors of patients with lung cancer. Nat. Med. 1996, 2, 985–991. [Google Scholar] [CrossRef]
- Chène, P. p53 as a drug target in cancer therapy. Expert Opin. Ther. Patents 2001, 11, 923–935. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Ganly, I.; Khuri, F.; Arseneau, J.; Kuhn, J.; Mccarty, T.; Landers, S.; Maples, P.; Romel, L.; Randlev, B.; et al. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: A phase II trial. Cancer Res. 2000, 60, 6359–6366. [Google Scholar]
- Boeckler, F.; Joerger, A.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [Green Version]
- Rippin, T.M.; Bykov, V.J.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Kuball, J.; Schuler, M.; Ferreira, E.A.; Herr, W.; Neumann, M.; Obenauer-Kutner, L.; Westreich, L.; Huber, C.; Wölfel, T.; Theobald, M. Generating p53-specific cytotoxic T lymphocytes by recombinant adenoviral vector-based vaccination in mice, but not man. Gene Ther. 2002, 9, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Svane, I.M.; Pedersen, A.E.; Johnsen, H.E.; Nielsen, D.; Kamby, C.; Gaarsdal, E.; Nikolajsen, K.; Claesson, M.H. Vaccination with p53-peptide? pulsed dendritic cells, of patients with advanced breast cancer: Report from a phase I study. Cancer Immunol. Immunother. 2004, 53, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Mu, Y.; Hallahan, D.E.; Lu, B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene 2004, 23, 7047–7052. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Cherton-Horvat, G.; Dragowska, V.; Baird, S.; Korneluk, R.G.; Durkin, J.P.; Mayer, L.D.; Lacasse, E.C. Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin. Cancer Res. 2003, 9, 2826–2836. [Google Scholar]
- Ohnishi, K.; Scuric, Z.; Schiestl, R.H.; Okamoto, N.; Takahashi, A.; Ohnishi, T. siRNA targeting NBS1 or XIAP increases radiation sensitivity of human cancer cells independent of TP53 status. Radiat. Res. 2006, 166, 454–462. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shiraki, K.; Fuke, H.; Inoue, T.; Miyashita, K.; Yamanaka, Y.; Saitou, Y.; Sugimoto, K.; Nakano, T. Targeting of X-linked inhibitor of apoptosis protein or survivin by short interfering RNAs sensitize hepatoma cells to TNF-related apoptosis-inducing ligand-and chemotherapeutic agent-induced cell death. Oncol. Rep. 2005, 14, 1311–1316. [Google Scholar] [CrossRef]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Investig. Dermatol. 1999, 113, 1076–1081. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.; Sen, S.; Muzio, L.L.; Mariggiò, M.A.; Singh, N. Antisense-mediated downregulation of anti-apoptotic proteins induces apoptosis and sensitizes head and neck squamous cell carcinoma cells to chemotherapy. Cancer Biol. Ther. 2005, 4, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.-X.; Zhang, H.-Y.; Gao, D.-X.; Wang, H.-Q.; Li, Y.-J.; Liu, G.-L. Antisurvivin oligonucleotides inhibit growth and induce apoptosis in human medullary thyroid carcinoma cells. Exp. Mol. Med. 2006, 38, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kami, K.; Doi, R.; Koizumi, M.; Toyoda, E.; Mori, T.; Ito, D.; Kawaguchi, Y.; Fujimoto, K.; Wada, M.; Miyatake, S.-I.; et al. Downregulation of survivin by siRNA diminishes radioresistance of pancreatic cancer cells. Surgery 2005, 138, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Quanxi, L.I.U.; Chaoying, D.O.N.G.; Lijun, L.I.; Jie, S.U.N.; Chunyun, L.I.; Liang, L.I. Inhibitory effects of the Survivin siRNA transfection on human lung adenocarcinoma cells SPCA1 and SH77. Chin. J. Lung Cancer 2011, 14, 18–22. [Google Scholar]
- Zhang, X.; Li, N.; Wang, Y.H.; Huang, Y.; Xu, N.Z.; Wu, L.Y. Effects of Survivin siRNA on growth, apoptosis and chemosensitivity of ovarian cancer cells SKOV3/DDP. Zhonghua Zhong liu za zhi Chin. J. Oncol. 2009, 31, 174–177. [Google Scholar]
- Yang, C.T.; Li, J.M.; Weng, H.H.; Li, Y.C.; Chen, H.C.; Chen, M.F. Adenovirus-mediated transfer of siRNA against survivin enhances the radiosensitivity of human non-small cell lung cancer cells. Cancer Gene Ther. 2010, 17, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Pennati, M.; Folini, M.; Zaffaroni, N. Targeting survivin in cancer therapy: Fulfilled promises and open questions. Carcinogenesis 2007, 28, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Liu, L.; Lu, J.; Qiu, S.; Yang, C.-Y.; Yi, H.; Wang, S. Cyclopeptide Smac mimetics as antagonists of IAP proteins. Bioorg. Med. Chem. Lett. 2010, 20, 3043–3046. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; McEachern, D.; Sun, H.; Bai, L.; Peng, Y.; Qiu, S.; Miller, R.; Liao, J.; Yi, H.; Liu, M.; et al. Therapeutic potential and molecular mechanism of a novel, potent, nonpeptide, Smac mimetic SM-164 in combination with TRAIL for cancer treatment. Mol. Cancer Ther. 2011, 10, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Rohn, J.L.; Noteborn, M.H.M. The viral death effector Apoptin reveals tumor-specific processes. Apoptosis 2004, 9, 315–322. [Google Scholar] [CrossRef]
- Philchenkov, A.; Zavelevich, M.; Kroczak, T.J.; Los, M. Caspases and cancer: Mechanisms of inactivation and new treatment modalities. Exp. Oncol. 2004, 26, 82–97. [Google Scholar]
- Yamabe, K.; Shimizu, S.; Ito, T.; Yoshioka, Y.; Nomura, M.; Narita, M.; Saito, I.; Kanegae, Y.; Matsuda, H. Cancer gene therapy using a pro-apoptotic gene, caspase-3. Gene Ther. 1999, 6, 1952–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cam, L.; Boucquey, A.; Coulomb-L’hermine, A.; Weber, A.; Horellou, P. Gene transfer of constitutively active caspase-3 induces apoptosis in a human hepatoma cell line. J. Gene Med. A Cross-Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2005, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, R.; Zou, X.; Gao, L.; Jin, H.; Du, R.; Xia, L.; Fan, D. Inhibitory effect of recombinant adenovirus carrying immunocaspase-3 on hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2007, 358, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis deregulation and the development of cancer multi-drug resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. U.S. National Library of Medicine. Available online: https://www.clinicaltrials.gov/ (accessed on 11 January 2022).
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Investig. 2007, 117, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Dömling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α−helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ding, C.; Zhang, J.; Xie, M.; Park, D.; Ding, Y.; Chen, G.; Zhang, G.; Gilbert-Ross, M.; Zhou, W.; et al. Modulation of Bax and mTOR for cancer therapeutics. Cancer Res. 2017, 77, 3001–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [PubMed] [Green Version]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Monian, P.; Jiang, X. Clearing the final hurdles to mitochondrial apoptosis: Regulation post cytochrome C release. Exp. Oncol. 2012, 34, 185–191. [Google Scholar]
- Amaravadi, R.K.; Schilder, R.J.; Martin, L.P.; Levin, M.; Graham, M.A.; Weng, D.E.; Adjei, A.A. A phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol. Cancer Ther. 2015, 14, 2569–2575. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Leblanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.-C.; Márk, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhász, E.; Pujol, J.-L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of Dulanermin in combination with Paclitaxel, Carboplatin, and Bevacizumab in advanced non–small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
- von Pawel, J.; Harvey, J.H.; Spigel, D.R.; Dediu, M.; Reck, M.; Cebotaru, C.L.; Humphreys, R.C.; Gribbin, M.J.; Fox, N.L.; Camidge, D.R. Phase II Trial of Mapatumumab, a fully human agonist monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with Paclitaxel and Carboplatin in patients with advanced non–small-cell lung cancer. Clin. Lung Cancer 2014, 15, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Attard, G.; Pacey, S.C.; Li, L.; Razak, A.; Perrett, R.; Barrett, M.; Judson, I.; Kaye, S.; Fox, N.L.; et al. Phase 1 and pharmacokinetic study of Lexatumumab in patients with advanced cancers. Clin. Cancer Res. 2007, 13, 6187–6194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camidge, D.R.; Herbst, R.S.; Gordon, M.S.; Eckhardt, S.G.; Kurzrock, R.; Durbin, B.; Ing, J.; Tohnya, T.M.; Sager, J.; Ashkenazi, A.; et al. A phase I safety and pharmacokinetic study of the death receptor 5 agonistic antibody PRO95780 in patients with advanced malignancies. Clin. Cancer Res. 2010, 16, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 2008, 7, 1001–1012. [Google Scholar] [CrossRef]
- Kline, C.L.B.; Ralff, M.D.; Lulla, A.R.; Wagner, J.M.; Abbosh, P.H.; Dicker, D.T.; Allen, J.E.; El-Deiry, W.S. Role of dopamine receptors in the anticancer activity of ONC201. Neoplasia 2017, 20, 80–91. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci. Transl. Med. 2013, 5, 71ra17. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. Targeting therapies for the p53 protein in cancer treatments. Annu. Rev. Cancer Biol. 2019, 3, 21–34. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53–MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Le Blanc, J.M.; Wang, C.; Zhan, T.; Zhuang, H.; Wang, P.; Yuan, Z.; Lu, B. Coadministration of Trametinib and Palbociclib radiosensitizes KRAS-mutant non–small cell lung cancers in vitro and in vivo. Clin. Cancer Res. 2016, 22, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Kim, S.R.; Lewis, J.M.; Cyrenne, B.M.; Monico, P.F.; Mirza, F.N.; Carlson, K.R.; Foss, F.M.; Girardi, M. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 2018, 9, 29193–29207. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Mondello, P.; Erazo, T.; Tannan, N.B.; Asgari, Z.; de Stanchina, E.; Nanjangud, G.; Seshan, V.E.; Wang, S.; Wendel, H.-G.; et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc. Natl. Acad. Sci. USA 2018, 115, 12034–12039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018, 133, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Bussenius, J.; Blazey, C.M.; Aay, N.; Anand, N.K.; Arcalas, A.; Baik, T.; Bowles, O.J.; Buhr, C.A.; Costanzo, S.; Curtis, J.K.; et al. Discovery of XL888: A novel tropane-derived small molecule inhibitor of HSP90. Bioorg. Med. Chem. Lett. 2012, 22, 5396–5404. [Google Scholar] [CrossRef]
- Park, M.A.; Zhang, G.; Martin, A.P.; Hamed, H.; Mitchell, C.; Hylemon, P.B.; Graf, M.; Rahmani, M.; Ryan, K.; Liu, X.; et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol. Ther. 2008, 7, 1648–1662. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caspases | Presence | Substrates | Functions |
|---|---|---|---|
| Caspase-1 | Spleen, liver, kidney, lung, heart | Lamins, Interleukins | Involved in inflammation, apoptosis induction when overexpressed [58] |
| Caspase-2 | Liver, CNS, kidney and lung development in embryo | Lamins, Golgin-160 | Apoptosis [59] |
| Caspase-3 | Broadly distributed | Caspases-6, -7, -9 | Apoptosis [60] |
| Caspase-4 | Lung, placenta, ovary, liver | Caspase-1 | Apoptosis [61] |
| Caspase-5 | Liver, lung | Max | Apoptosis, inflammation [62] |
| Caspase-6 | Liver, lung, skeletal muscle | PARP, caspase-3, NuMA, lamins, FAK, keratin-18 | Apoptosis [63] |
| Caspase-7 | Lung, kidney, liver, heart, spleen, testis | PARP, GAS2, EMAP II, calpastatin, FAK | Apoptosis [64] |
| Caspase-8 | Leukocytes, thymus, spleen, liver | Caspases-3, -4, -6, -7, -9, -10, -13 | Apoptosis [65] |
| Caspase-9 | Heart, liver, skeletal muscle, pancreas | Caspase-3, PARP, procaspase-9, caspase-7 | Apoptosis [66] |
| Caspase-10 | Tissues | Caspases-3, -4, -6, -7, -8, -9 | Apoptosis [67] |
| Caspase-11 | Brain microglia | Caspases-3, -1 | Apoptosis, inflammation [68] |
| Caspase-12 | Endoplasmic reticulum (ER) | Caspases-1, -4, -5, -11 | Apoptosis-mediated by ER stress [69] |
| Caspase-13 | Lymphocytes, placenta, spleen | Caspase-8 | Inflammation [69] |
| Caspase-14 | Epidermal cells | Caspases-8, -10 | Inflammation [70] |
| BCL-2 Protein | Location | Roles | Refs |
|---|---|---|---|
| BAX | Cytosol | Liberation of apoptogenic factors and induction of caspases | [98] |
| BAK | Integral mitochondrial membrane protein | Conformational changes in BAK take place to form larger complexes in apoptosis and create pores in the mitochondrial membrane to liberate apoptogenic factors to promote apoptosis | [99] |
| BID | Cytosol and membrane | Directly activate BAX | [100] |
| BCL-2 | Mitochondria, nucleus, endoplasmic reticulum | Prevents apoptosis by maintaining integrity of mitochondrial membrane integrity | [101,102] |
| BCL-XL | Mitochondrial transmembrane | Prevents release of cytochrome c via mitochondrial pore, thereby inhibiting activation of caspases by cytochrome c | [102] |
| MCL-1 | Nucleus, mitochondria | Associated with BAK1, BCL-2-associated death promoter, NOXA, BCL2L11 and PCNA | [103,104] |
| BCL-w/BCL2L2 | Mitochondrion | Under cytotoxic conditions downregulate apoptosis | [105] |
| A1/BFL-1 | Mitochondria, nucleus | unknown | [106,107] |
| BIM/BCL2L11 | Mitochondria | Interacts with BCL-2 or BCL-XL and prevents their anti-apoptotic actions | [108] |
| PUMA | Mitochondria | unknown; regulated by p53 transcriptionally | [109,110] |
| BAD | Mitochondria | Generate a complex with BCL-2 and BCL-XL, inhibits them, thereby promoting BAX/BAK-mediated apoptosis | [111] |
| BIK/BLK | Endoplasmic reticulum | unknown | [112] |
| NOXA/PMAIP1 | Mitochondria | unknown | [47,113] |
| BMF | Mitochondria | unknown | [114] |
| Treatment | Remarks | Refs |
|---|---|---|
| Attacking the BCL-2 family | ||
| Oblimersen sodium | Showed chemosensitivity along with anticancer drugs with significant improvement in myeloid leukemia | [123,124] |
| BCL-2 family inhibitors (Small molecule) | Sodium butyrate, fenretinide, depsipetide and flavipirodo are known to alter gene or protein expression. While ABT-263, GX15-070, ABT-737, HA14-1 and gossypol, affect the proteins directly | [125] |
| BH3 mimetics | ABT-737 inhibit anti-apoptotic proteins namely BCL-2, BCL-XL and BCL-w | [126] |
| ATF4, ATF3 and NOXA prevent MCL-1 functioning | [127] | |
| Suppressing the Bcl family anti-apoptotic proteins/genes | BCL-2 specific siRNA prevent target gene expression and promote anti-proliferation and pro-apoptotic activity in pancreatic cancer cells | [128] |
| Suppressing BMI-1 is known to decrease the expression of pAKT and BCL-2, it makes them sensitive to doxorubicin | [129] | |
| Targeting p53 | ||
| p53-based gene therapy | Wild-type p53 genes having retroviral vector introduced into cancer cells showed significant improvement | [130] |
| Introduction of wild type p53 gene makes head and neck tumor cells, and prostate cancers sensitive to radiotherapy | [131] | |
| ONYX-015 can disrupt tumor cells deficient in p53 | [132] | |
| p53-dependent drug therapy | ||
| Small molecules | PhiKan083 (A24275) binds and restores mutant p53 | [133] |
| CP-31398 inserted with DNA and disrupts the DNA-p53 complex, leading to restoration of unstable p53 mutants | [134] | |
| Other agents | Nutlins disrupt MSM2-p53 interaction, provide stability to p53 and promote death in cancer cells | [135] |
| MI-219 breaks MDM2-p53 interaction, leading to inhibition of cell multiplication and the promotion of apoptosis in cancer cells | [136] | |
| Tenovins reduce tumor growth in vivo | [137] | |
| p53-based immunotherapy | Vaccine having recombinant replication-defective adenoviral vector in combination with human wild-type p53 showed improvement | [138] |
| p53-specific T cell responses seen when given p53 peptide | [139] | |
| Targeting inhibitors of apoptosis proteins (IAPs) | ||
| Targeting XIAP by antisense approach | Improved tumor control by radiotherapy | [140] |
| Antisense oligonucleotides increase chemotherapeutic activity | [141] | |
| Targeting XIAP by siRNA approach | siRNAs targeting XIAP promote enhanced sensitivity towards radiotherapy | [142] |
| siRNAs targeting XIAP make hepatoma cells sensitive towards death receptor and chemotherapy | [143] | |
| Targeting survivin by antisense approach | Transfection of anti-sense survivin into melanoma cells promotes apoptosis | [144] |
| Promote apoptosis and sensitivity of cancer cells towards chemotherapy | [145] | |
| Prevent growth of thyroid carcinoma cells | [146] | |
| Targeting survivin by siRNA approach | Decrease survivin expression and lower the resistance to radiotherapy in pancreatic cancer cells | [147] |
| Prevent proliferation and promote apoptosis in lung adenocarcinoma cells | [148] | |
| Downregulate survivin expression, prevent multiplication and increase apoptosis in ovarian cancer | [149] | |
| Increase radiosensitivity in cancer cells | [150] | |
| IAP antagonists (Small molecules antagonists) | Hsp90 inhibitors and Cyclin-dependent kinase inhibitors are reported to target survivin | [151] |
| Cyclopeptide SMAC mimetics 2 and 3 attaches to XIAP and cIAP-1/2, thereby promoting the induction of caspases- 9 and -3/-7 | [152] | |
| SM-164 increases TRAIL functioning | [153] | |
| Targeting caspases | ||
| Caspase-dependent drug therapy | Apoptin promotes apoptosis in malignant cells | [154] |
| Small molecule caspase activators stimulate caspase, promoting enhanced drug sensitivity in tumor cells | [155] | |
| Caspase-dependent gene therapy | Caspase-3 gene therapy is reported to promote induction of extensive apoptosis | [156] |
| Caspase-3 gene introduction into Huh7 human hepatoma cells promotes apoptosis | [157] | |
| Immunocaspase-3 in a recombinant adenovirus showed anticancer effect in hepatocellular cancer | [158] | |
| Target | Clinical Trial | Histology | Trial Identity ** | |
|---|---|---|---|---|
| Dual BCL-2 and BCL-XL inhibitors | Navitoclax | YES (Phase I/II) | CLL, melanoma, solid tumors | NCT02079740, NCT02143401, NCT01989585, NCT02520778 |
| APG-1252 | YES (Phase I/II) | SCLC, solid tumors | NCT03387332 | |
| AZD4320 | No | Childhood ALL | - | |
| S44563 | No | Melanoma, SCLC, | - | |
| BCL2–32 | No | NHL | - | |
| BM-1197 | No | Colorectal cancer | - | |
| Selective BCL-2 inhibitors | Venetoclax | Yes (Phase I-III) | CLL, AML | - |
| S55746 (BCL201) | Yes (Phase I) | NHL, multiple myeloma | NCT02603445, NCT02920697 | |
| APG-2575 | Yes (Phase I) | NHL, AML | NCT03537482, NCT03913949 | |
| BCL-XL inhibitors | ABBV-155 * | Yes (Phase I) | Solid tumors | NCT03595059 |
| WEHI-539 | No | Breast cancer | - | |
| A-1155463 | No | AML | - | |
| A-1331852 | No | Soft-tissue sarcoma | - | |
| MCL-1 inhibitors | AMG 176 | Yes (Phase I) | NHL, AML | NCT02675452, NCT03797261 |
| MIK665 (S64315) | Yes (Phase I) | NHL, AML | NCT02992483, NCT03672695, NCT02979366 | |
| AZD5991 | Yes (Phase I) | NHL, AML | NCT03218683 | |
| S63845 | No | NHL, AML | - | |
| UMI-77 | No | Pancreatic cancer | - | |
| A-1210477 | No | Esophageal carcinoma | - | |
| VU661013 | No | AML | - | |
| IAP inhibitors and SMAC mimetic antagonists | LCL161 | Yes (Phase I/II) | Colorectal cancer, multiple myeloma, Polycythemia vera, myelofibrosis | NCT02649673, NCT02098161, NCT03111992 |
| Birinapant (TL32711) | Yes (Phase I/II) | Advanced solid tumors, NHL | NCT03803774, NCT02587962 |
| Target | Clinical Trials | Cancer | Trial Identity * |
|---|---|---|---|
| Death Receptor Agonists (DR4/5) | |||
| GEN1029 | Yes (Phase I) | Colorectal cancer, renal carcinoma, triple negative breast cancer, pancreatic cancer, gastric cancer | NCT03576131 |
| ABBV-621 | Yes (Phase I) | AML, NHL, pancreatic cancer | NCT03082209 |
| MM-201 | No | Sarcoma | - |
| TLY012 | No | Fibrosis | - |
| Approaches targeting p53 | |||
| Idasanutlin (RG73882) | Yes (Phase I) | Breast cancer, AML, NHL, multiple myeloma | NCT03850535, NCT02670044, NCT02545283, NCT02633059, NCT03566485, NCT03135262 |
| AMG-232 | Yes (Phase I/II) | AML, multiple myeloma, sarcoma | NCT03041688, NCT03217266, NCT03031730 |
| HDM201 | Yes (Phase I) | AML | NCT03940352 |
| APG-115 | Yes (Phase I) | Advanced solid tumors, AML, melanoma | NCT02935907, NCT03611868, NCT03781986 |
| DS-3032b | Yes (Phase I) | AML, solid tumors | NCT03634228, NCT02319369, NCT01877382 |
| BI 907828 | Yes (Phase I) | Solid tumors | NCT03449381 |
| ALRN-6924 *** | Yes (Phase I) | Solid tumors | NCT03725436 |
| Restore wild type activity of mutant p53 | |||
| APR246 | Yes (Phase I) | AML, esophageal carcinoma, ovarian cancer, melanoma | NCT02999893, NCT02098343, NCT03588078, NCT03391050, NCT03391050 |
| Other cell death mechanisms associated with apoptosis | |||
| ONC201 ** | Yes (Phase I/II) | NHL, breast cancer, multiple myeloma, colorectal cancer, endometrial cancer, AML | NCT03099499, NCT02863991, NCT03416530, NCT02420795, NCT03394027, NCT03295396, NCT03791398, NCT02392572 |
| Epigenetic modulators for stimulating intrinsic pathway of apoptosis | |||
| Fimepinostat +venetoclax | Yes (Phase I/II) | NHL | NCT01742988 |
| Azacytidine or decitabine + venetoclax | Yes (Phase I-III) | AML | NCT03404193, NCT03941964 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, V.; Khurana, A.; Navik, U.; Allawadhi, P.; Bharani, K.K.; Weiskirchen, R. Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics. Sci 2022, 4, 15. https://doi.org/10.3390/sci4020015
Singh V, Khurana A, Navik U, Allawadhi P, Bharani KK, Weiskirchen R. Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics. Sci. 2022; 4(2):15. https://doi.org/10.3390/sci4020015
Chicago/Turabian StyleSingh, Vishakha, Amit Khurana, Umashanker Navik, Prince Allawadhi, Kala Kumar Bharani, and Ralf Weiskirchen. 2022. "Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics" Sci 4, no. 2: 15. https://doi.org/10.3390/sci4020015
APA StyleSingh, V., Khurana, A., Navik, U., Allawadhi, P., Bharani, K. K., & Weiskirchen, R. (2022). Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics. Sci, 4(2), 15. https://doi.org/10.3390/sci4020015