Development of a Low-Density DNA Microarray for Detecting Tick-Borne Bacterial and Piroplasmid Pathogens in African Cattle

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Origin, DNA Extraction, PCR and Sanger Sequencing
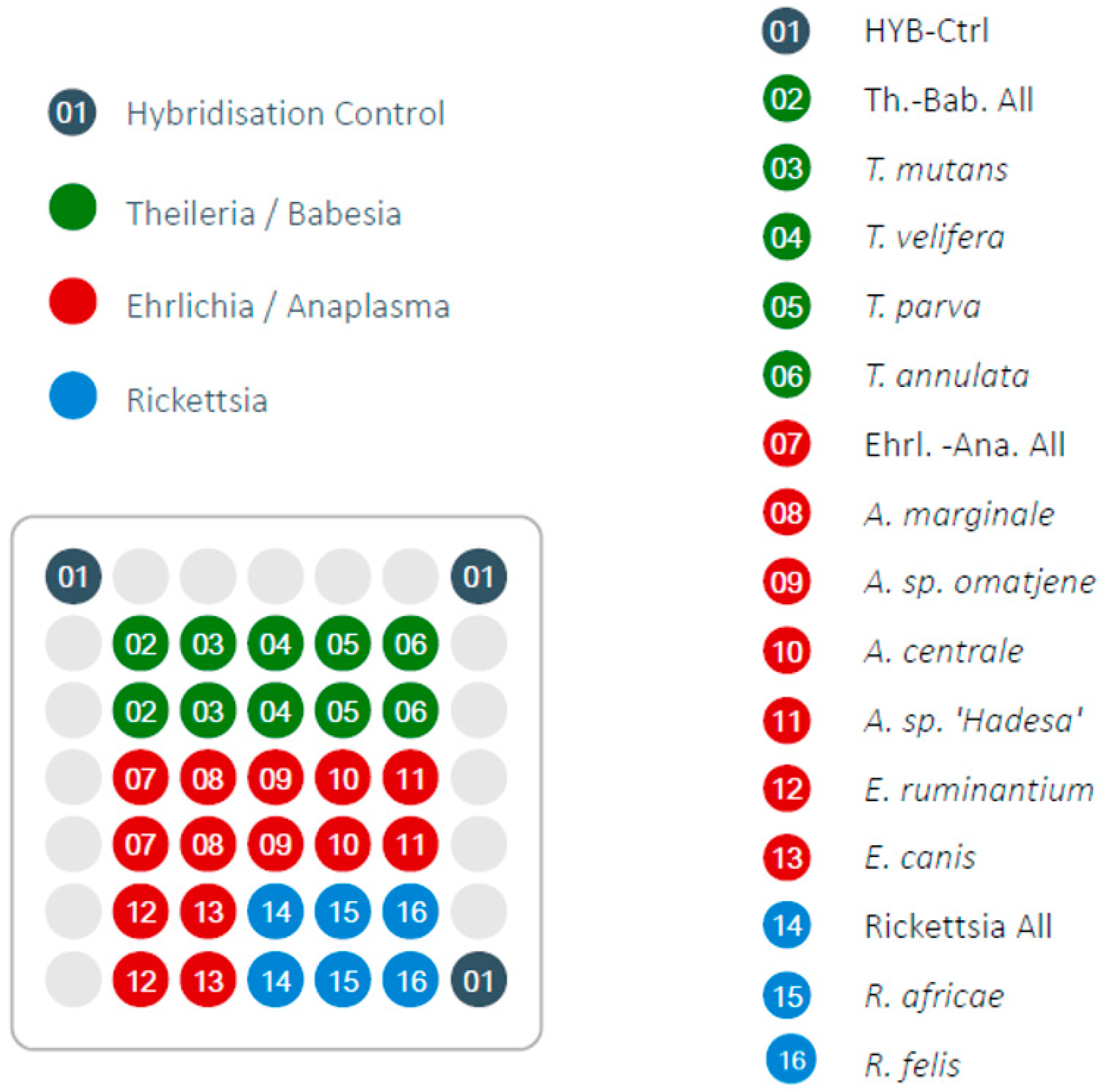
2.2. LCD-Array Specification and Validation
2.3. LCD-Array Workflow
3. Results
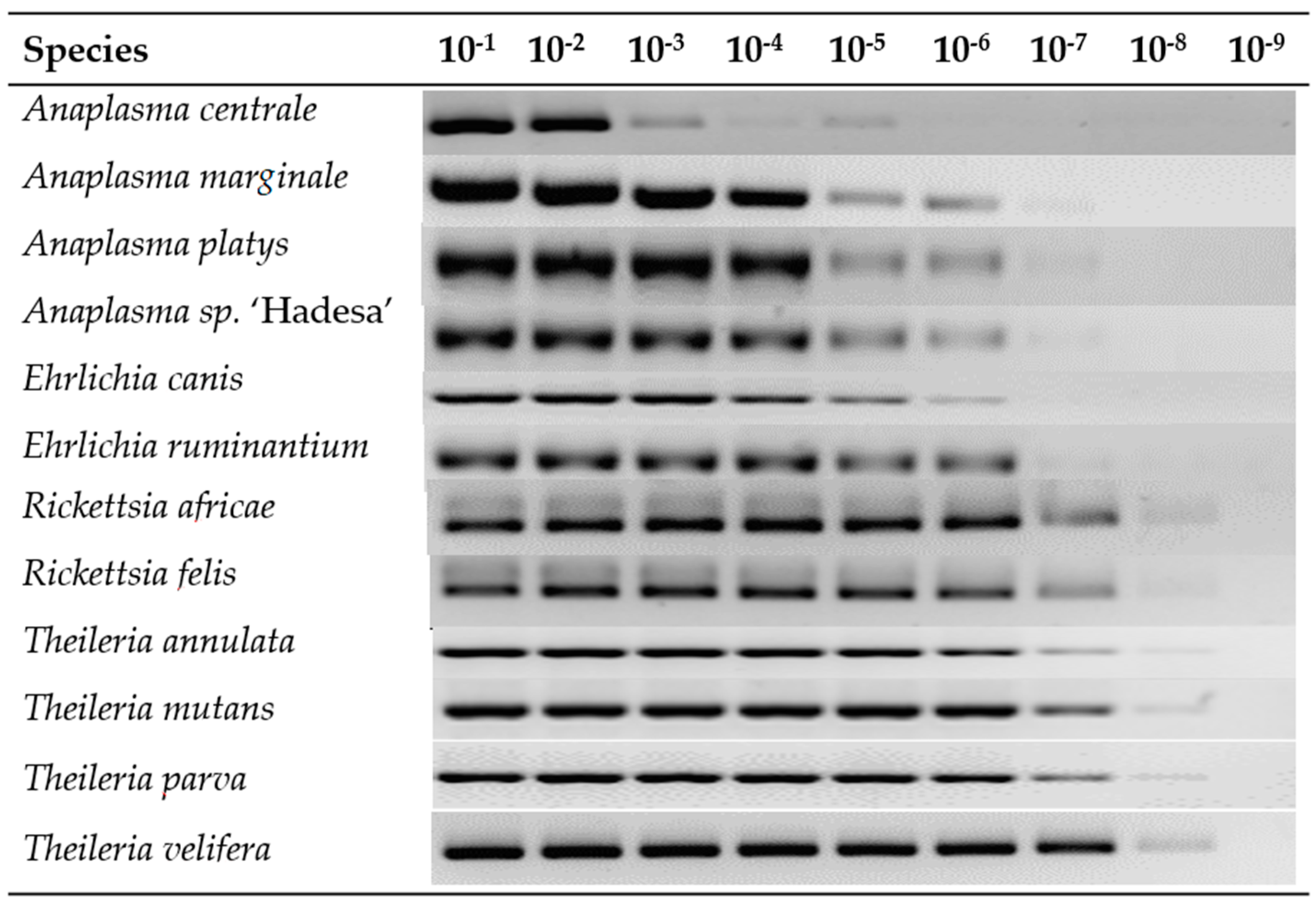
3.1. LCD-Array Performance of Synthetic Inserts (Plasmids)
3.2. LCD-Array Performance of Cattle Blood Samples from North Cameroon
3.2.1. Anaplasma
3.2.2. Ehrlichia
3.2.3. Rickettsia
3.2.4. Babesia
3.2.5. Theileria
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lorusso, V.; Wijnveld, M.; Majekodunmi, A.O.; Dongkum, C.; Fajinmi, A.; Dogo, A.G.; Thrusfield, M.; Mugenyi, A.; Vaumourin, E.; Igweh, A.C.; et al. Tick-borne pathogens of zoonotic and veterinary importance in Nigerian cattle. Parasit Vectors 2016, 9, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Mamoudou, A.; Nguetoum, C.; Sevidzem, L.; Manchang, K.; Njongui, J.; Zoli, P. Bovine babesiosis and anaplasmosis in some cattle farms in the Vina division. Int. J. Livest. Res. 2017, 7, 69–80. [Google Scholar] [CrossRef]
- Bell-Sakyi, L.; Koney, E.B.M.; Dogbey, O.; Walker, A.R. Incidence and prevalence of tick-borne haemoparasites in domestic ruminants in Ghana. Vet. Parasitol. 2004, 124, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Spickler, A.R. Zoonotic Species Ehrlichiosis and Anaplasmosis. In Ehrlichiosis and Anaplasmosis; Technical Report for Iowa State University Center for Food Security and Public Health: Ames, IA, USA, 2013; pp. 1–14. [Google Scholar]
- Parola, P.; Paddock, C.D.; Socolovschi, C.; Labruna, M.B.; Mediannikov, O.; Kernif, T.; Abdad, M.Y.; Stenos, J.; Bitam, I.; Fournier, P.E.; et al. Update on tick-borne rickettsioses around the world: A geographic approach. Clin. Microbiol. Rev. 2013, 26, 657–702. [Google Scholar] [CrossRef] [PubMed]
- Awa, D.N. Serological survey of heartwater relative to the distribution of the vector Amblyomma variegatum and other tick species in north Cameroon. Vet. Parasitol. 1997, 68, 165–173. [Google Scholar] [CrossRef]
- Molad, T.; Mazuz, M.L.; Fleiderovitz, L.; Fish, L.; Savitsky, I.; Krigel, Y. Molecular and serological detection of A. centrale and A. marginale infected cattle grazing within an endemic area. Vet. Microbiol. 2006, 113, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.B.; Andre, M.R.; Machado, R.Z. Low genetic diversity of Anaplasma marginale in calves in an endemic area for bovine anaplasmosis in the state of Sao Paulo, Brazil. Ticks Tick Borne Dis. 2016, 7, 20–25. [Google Scholar] [CrossRef]
- Mans, B.J.; Pienaar, R.; Latif, A.A. A review of Theileria diagnostics and epidemiology. Int. J. Parasitol. Parasites Wildl. 2015, 4, 104–118. [Google Scholar] [CrossRef]
- Nijhof, A.M.; Bodaan, C.; Postigo, M.; Nieuwenhuijs, H.; Opsteegh, M.; Franssen, L.; Jebbink, F.; Jongejan, F. Ticks and Associated pathogens collected from domestic animals in the Netherlands. Vector-Borne Zoonotic Dis. 2007, 7, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.I.; Somasekar, S.; Naccache, S.N.; Bandeira, A.C.; Tauro, L.B.; Campos, G.S.; Chiu, Y.C. Coinfections of Zika and Chikungunya Viruses in Bahia, Brazil, Identified by Metagenomic Next-Generation Sequencing. J. Clin. Microbiol. 2016, 54, 2348–2353. [Google Scholar] [CrossRef] [Green Version]
- Beltramo, C.; Riina, M.V.; Colussi, S.; Campia, V.; Maniaci, M.G.; Biolatti, C.; Trisorio, S.; Modesto, P.; Peletto, S.; Acutis, P.L. Validation of a DNA biochip for species identification in food forensic science. Food Control. 2017, 78, 366–373. [Google Scholar] [CrossRef]
- Hailemariam, Z.; Krücken, J.; Baumann, M.; Ahmed, J.S.; Clausen, P.H.; Nijhof, A.M. Molecular detection of tick-borne pathogens in cattle from Southwestern Ethiopia. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, R.; Paweska, J.T.; Petersen, N.; Grobbelaar, A.A.; Leman, A.P.; Hewson, R.; Georges-Courbot, M.C.; Papa, A.; Heiser, V.; Panning, M.; et al. Low-density macroarray for rapid detection and identification of Crimean-Congo hemorrhagic fever virus. J. Clin. Microbiol. 2009, 47, 1025–1030. [Google Scholar] [CrossRef]
- Chang, C.I.; Hung, P.H.; Wu, C.C.; Cheng, T.C.; Tsai, J.M.; Lin, K.J.; Lin, C.Y. Simultaneous detection of multiple fish pathogens using a naked-eye readable DNA microarray. Sensors 2012, 12, 2710–2728. [Google Scholar] [CrossRef] [PubMed]
- Szafrański, S.P.; Deng, Z.L.; Tomasch, J.; Jarek, M.; Bhuju, S.; Meisinger, C. Functional biomarkers for chronic periodontitis and insights into the roles of Prevotella nigrescens and Fusobacterium nucleatum; a metatranscriptome analysis. Biofilms Microbi. 2015, 15, 2055–5008. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Bettstetter, M.; Becher, A.; Lessel, M.; Bank, C.; Krams, M.; Becker, I.; Hartmann, A.; Jagla, W.; Gaumann, A. Shift in prevalence of HPV types in cervical cytology specimens in the era of HPV vaccination. Oncol. Lett. 2016, 12, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, A.; von Bomhard, W.; Antweiler, E.; Tintelnot, K. Molecular identification of fungal pathogens in nodular skin lesions of cats. Med. Mycol. 2015, 53, 132–144. [Google Scholar] [CrossRef] [PubMed]
- El-Ashker, M.; Hotzel, H.; Gwida, M.; El-Beskawy, M.; Silaghi, C.; Tomaso, H. Molecular biological identification of Babesia, Theileria, and Anaplasma species in cattle in Egypt using PCR assays, gene sequence analysis and a novel DNA microarray. Vet. Parasitol. 2015, 207, 329–334. [Google Scholar] [CrossRef]
- Nijhof, A.M.; Penzhorn, B.L.; Lynen, G.; Mollel, J.O.; Morkel, P.; Bekker, C.P.J.; Cornelis, P.J.; Jongejan, F. Babesia bicornis sp. nov. and Theileria bicornis sp. nov.: Tick-borne parasites associated with mortality in the Black Rhinoceros (Diceros bicornis). J. Clin. Microbiol. 2003, 41, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Omar Abdallah, M.; Niu, Q.; Yu, P.; Guan, G.; Yang, J.; Chen, Z.; Liu, G.; Wei, Y.; Luo, J.; Yin, H. Identification of piroplasm infection in questing ticks by RLB: A broad range extension of tick-borne piroplasm in China? Parasitol. Res. 2016, 115, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Neitz, W.O. A consolidation of our knowledge of the transmission of tick-borne disease. Onderstepoort J. Vet. Res. 1956, 27, 115–163. [Google Scholar]
- Oryan, A.; Namazi, F.; Sharifiyazdi, H.; Razavi, M.; Shahriari, R. Clinicopathological findings of a natural outbreak of Theileria annulata in cattle: An emerging disease in Southern Iran. Parasitol. Res. 2013, 112, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.S.; Kandil, O.M.; Nasr, S.M.; Hendawy, S.H.M.; Habeeb, S.M.; Mabrouk, D.M.; Silva, M.G.; Suarez, C.E. Serological and molecular diagnostic surveys combined with examining hematological profiles suggests increased levels of infection and hematological response of cattle to babesiosis infections compared to native buffaloes in Egypt. Parasit Vectors 2015, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bażanów, B.A.; Pacoń, J.; Gadzała, Ł.; Frącka, A.; Welz, M.; Paweska, J. Vector and serologic survey for Crimean–Congo Hemorrhagic Fever Virus in Poland. Vector-Borne Zoonotic Dis. 2017, 17, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Ah Tow, L.; Cowan, D.A. Non-specificity of Staphylococcus generic primers. Microbiology 2003, 149, 1605–1607. [Google Scholar] [CrossRef]
- Jain, N.K.; Roy, I. Trehalose and protein stability. Curr. Protoc. Protein Sci. 2010, 59, 1–12. [Google Scholar] [CrossRef]
- Call, D.R. Challenges and opportunities for pathogen detection using DNA microarrays. Crit. Rev. Microbiol. 2005, 31, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hilario, E.; Mackay, J. Microarrays. In Protocols for Nucleic Acid Analysis by Nonradioactive Probes; Humana Press: New York, NY, USA, 2007; pp. 265–300. [Google Scholar] [CrossRef]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Gene Target | Primer Sequence | Annealing Temp. | Amplicon Size [bp] | Reference |
|---|---|---|---|---|---|
| Babesia/Theileria | 18S rRNA | GAC ACA GGG AGG TAG TGA CAA G | 57 °C | 460–500 | [20] |
| b-CTA AGA ATT TCA CCT CTG ACA GT | |||||
| Anaplasma/Ehrlichia | 16S rRNA | AGA GTT TGA TCM TGG YTC AGA A | 55 °C | 460–520 | This study |
| b-GAG TTT GCC GGG ACT TYT TC | |||||
| Rickettsia | 16S rRNA | GAA CGC TAT CGG TAT GCT TAA CAC A | 64 °C | 350–400 | [10] |
| b-CAT CAC TCA CTC GGT ATT GCT GGA |
| Species | Copies/µL Pre-PCR * | LOD Post-PCR * | LOD LCD-Array |
|---|---|---|---|
| Anaplasma centrale | 75 | 10−5 | 10−8 |
| Anaplasma marginale | 31 | 10−7 | 10−8 |
| Anaplasma platys | 28 | 10−7 | 10−8 |
| Anaplasma sp. ‘Hadesa’ | 34 | 10−7 | 10−8 |
| Ehrlichia canis | 60 | 10−6 | 10−8 |
| Ehrlichia ruminantium | 40 | 10−7 | 10−8 |
| Rickettsia africae | 3 | 10−8 | 10−9 |
| Rickettsia felis | 2 | 10−8 | 10−9 |
| Theileria annulata | 6 | 10−8 | 10−9 |
| Theileria mutans | 3 | 10−8 | 10−9 |
| Theileria parva | 7 | 10−8 | 10−9 |
| Theileria velifera | 1 | 10−8 | 10−9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abanda, B.; Paguem, A.; Achukwi, M.D.; Renz, A.; Eisenbarth, A. Development of a Low-Density DNA Microarray for Detecting Tick-Borne Bacterial and Piroplasmid Pathogens in African Cattle. Trop. Med. Infect. Dis. 2019, 4, 64. https://doi.org/10.3390/tropicalmed4020064
Abanda B, Paguem A, Achukwi MD, Renz A, Eisenbarth A. Development of a Low-Density DNA Microarray for Detecting Tick-Borne Bacterial and Piroplasmid Pathogens in African Cattle. Tropical Medicine and Infectious Disease. 2019; 4(2):64. https://doi.org/10.3390/tropicalmed4020064
Chicago/Turabian StyleAbanda, Babette, Archile Paguem, Mbunkah Daniel Achukwi, Alfons Renz, and Albert Eisenbarth. 2019. "Development of a Low-Density DNA Microarray for Detecting Tick-Borne Bacterial and Piroplasmid Pathogens in African Cattle" Tropical Medicine and Infectious Disease 4, no. 2: 64. https://doi.org/10.3390/tropicalmed4020064
APA StyleAbanda, B., Paguem, A., Achukwi, M. D., Renz, A., & Eisenbarth, A. (2019). Development of a Low-Density DNA Microarray for Detecting Tick-Borne Bacterial and Piroplasmid Pathogens in African Cattle. Tropical Medicine and Infectious Disease, 4(2), 64. https://doi.org/10.3390/tropicalmed4020064