
Densification of Ceramic Matrix Composite Preforms by Si2N2O Formed by Reaction of Si with SiO2 under High Nitrogen Pressure. Part 1: Materials Synthesis


Abstract
:1. Introduction
1.1. Ceramic Matrix Composites
1.2. Choice of the Ceramic Matrix
1.3. Si-O-N Chemical System
1.3.1. Phase Formation and Phase Stability
1.3.2. Reaction Pathways
Reactions Involving Solid Silicon
Si3N4 Formed by Nitriding Solid Silicon
Reactions Involving Liquid Silicon
Si2N2O Formed in the Presence of Liquid Silicon
2. Materials and Methods
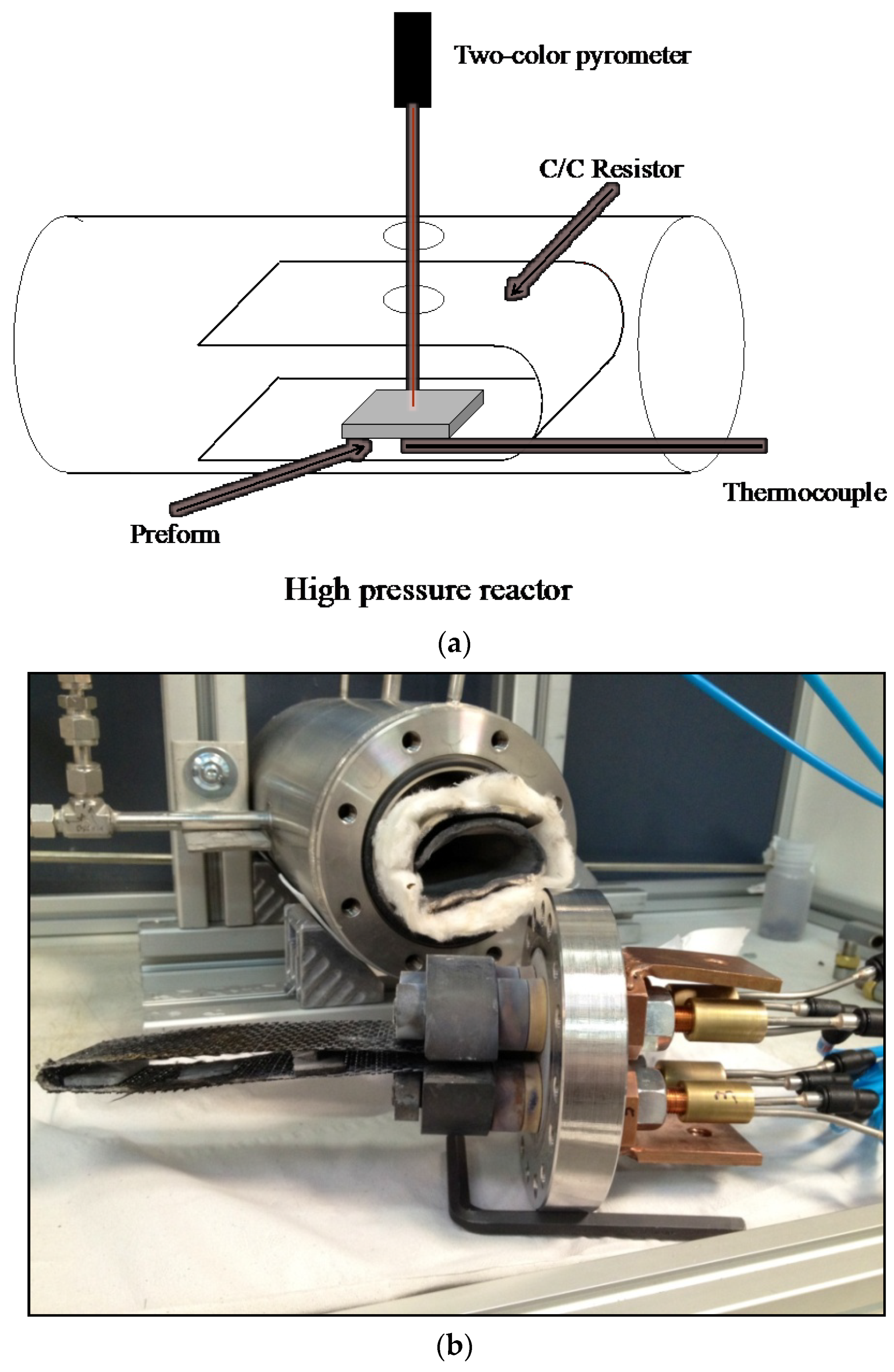
2.1. Reactor
2.2. Measurement of the Thermal Events Associated with the Chemical Reactions Occurring Inside the Preforms
2.3. Experimental Procedure
2.4. Nature and Preparation of the 3D Preform
2.5. Powder and Slurry Preparations
- -
- Aldrich Silicon powder, 325 mesh, purity 99% (trace of metals, nature not specified)
- -
- Aldrich Silicon dioxide with a quartz structure, 0.5–10 μm grain size (80% in the range 1–5 μm), purity 99% (trace of metals, nature not specified)
2.6. Powder Impregnation of the Preform
2.7. Silica Powder Produced by Oxidation of Silicon Powder Inside the Preform
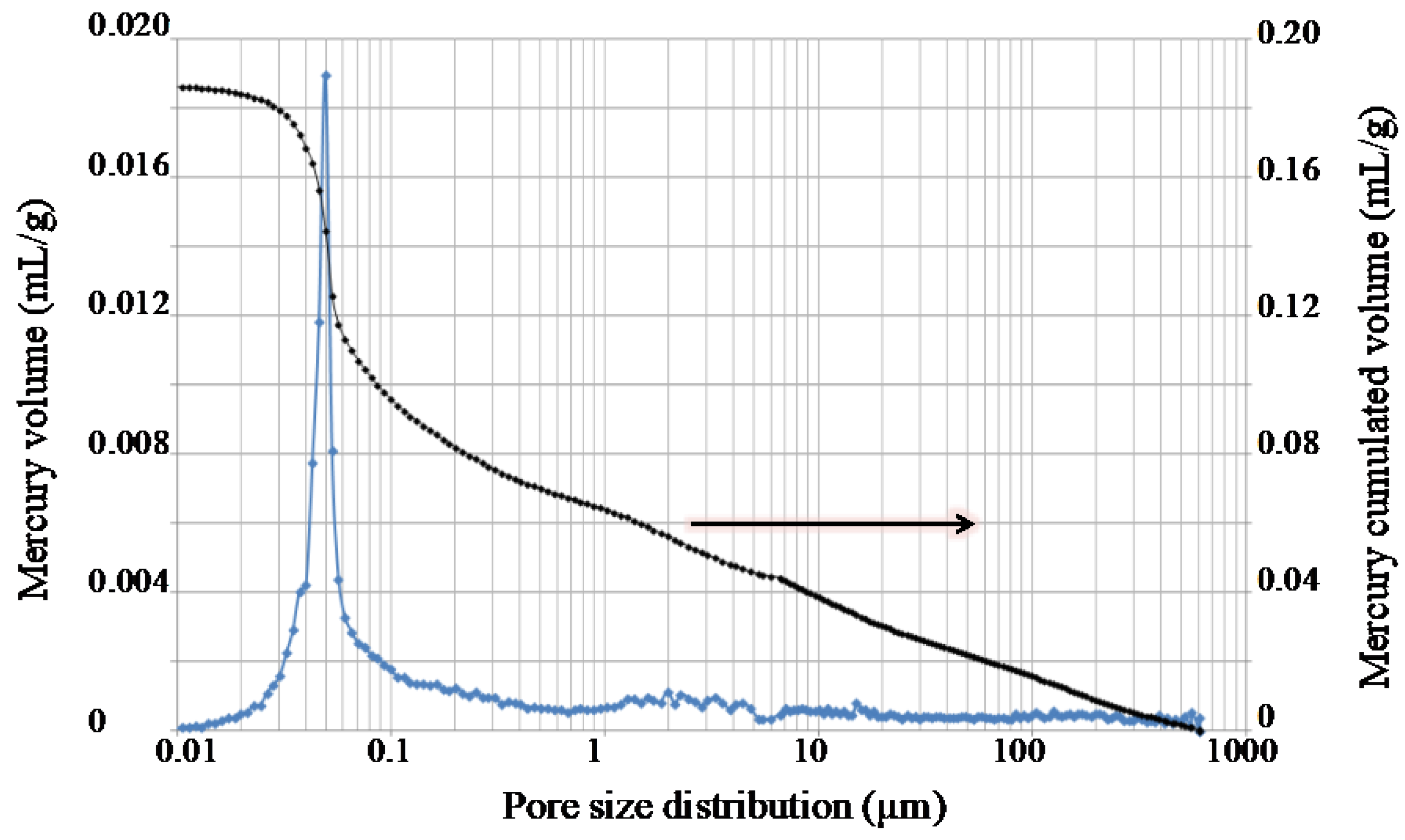
2.8. Characterization of the Processed Composites
3. Results
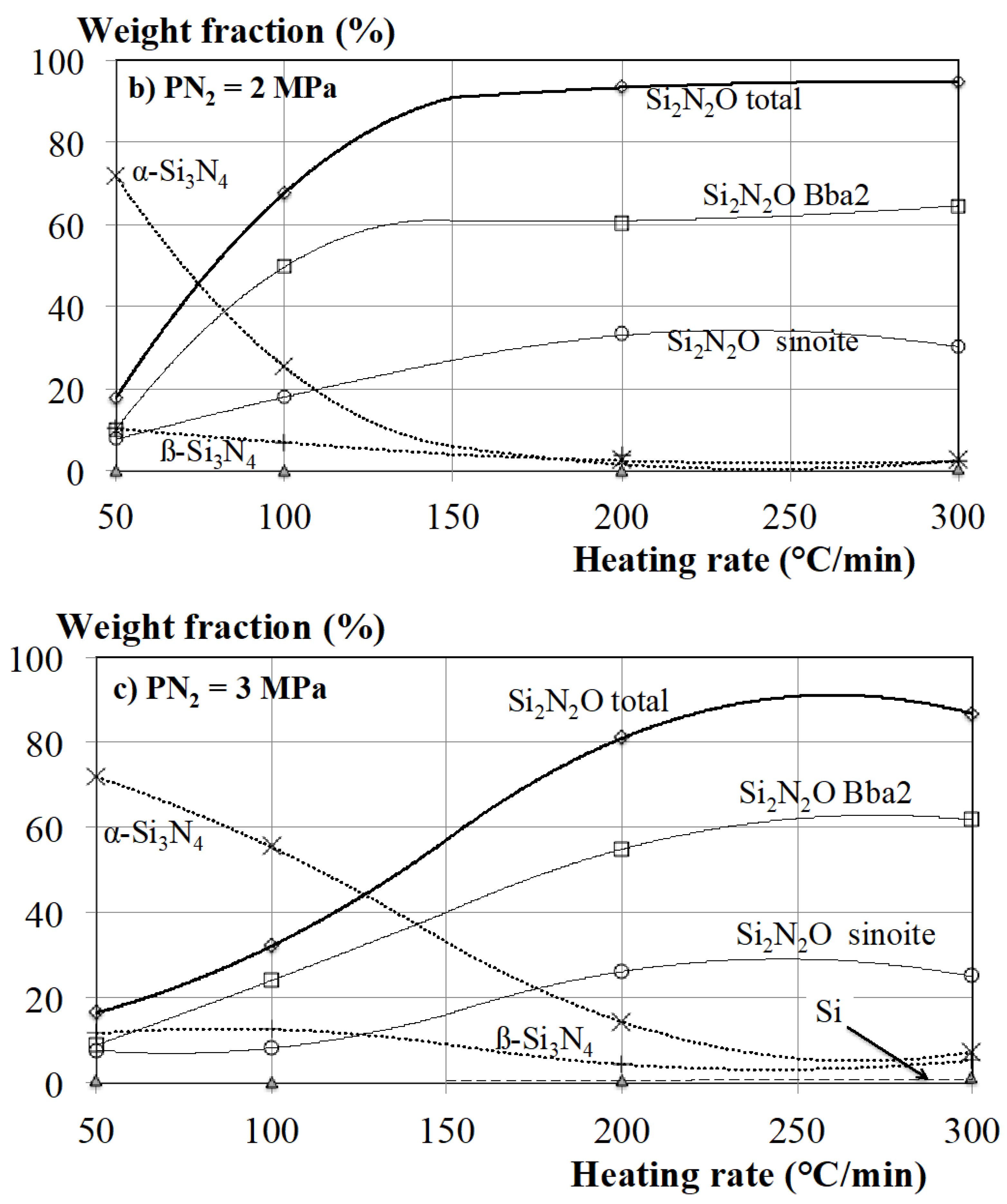
3.1. Effect of Both the Nitrogen Pressure and Heating Rate of the Sample on the Composition of the Processed Material
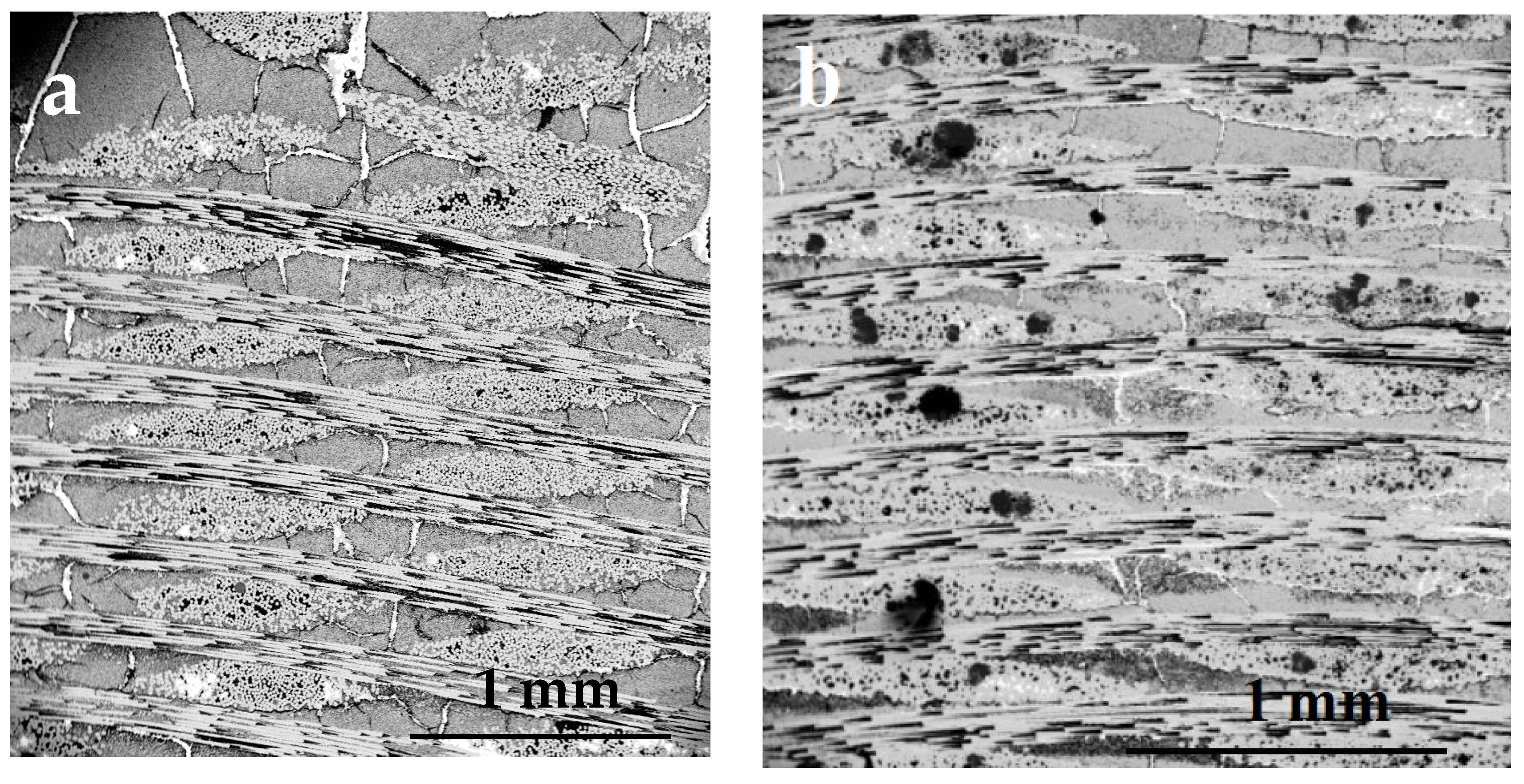
3.2. SEM Observation of the Processed Composites
3.3. Study of the Thermal Peaks Measured by Optical Pyrometry
3.4. Modified Process Involving Silica Produced by Oxidation of Silicon inside the Preform
3.5. Influence of the Thickness of the SiC Consolidation Layer
4. Discussion
- The formation of α-Si3N4 and ß-Si3N4 should be considered as independent of each other.
- Two different pathways, both of which require the presence of liquid Si and a dissolution process, can contribute to the formation of Si2N2O:
- ○
- A reaction involving SiO2 in contact with liquid silicon including dissolved N atoms.
- ○
- A reaction involving Si3N4 in contact with liquid silicon including dissolved O atoms coming from SiO2 also in contact with liquid Si. However, the presence of high nitrogen pressures stabilizes Si3N4 and tends to inhibit this reaction.
5. Conclusions
- Two allotropic forms of Si2N2O, as evidenced by XRD, are formed during the reactions occurring inside the preform: the well-known sinoite form and an additional new form of Si2N2O isostructural with the already-described Bba2 Si1.8Al0.2O1.2N1.8 phase, but without including aluminum. This second Si2N2O form is the main phase formed in all the experiments carried out in this study.
- At low heating rate (50 °C/min), the reaction of formation of Si3N4 and that of Si2N2O are successive and separated in time and, thus, in temperatures. The first reaction observed when increasing temperature is the formation of Si3N4, while silicon is still in the solid state: both the formation of α-Si3N4 by reaction between nitrogen and gaseous silicon and formation of ß-Si3N4 by reaction between nitrogen and solid silicon. Both reactions stop after a while, when the Si grains are entirely covered by Si3N4. The formation of Si2N2O occurs at a higher temperature, when the silicon grains begin to melt. It is observed that, when the two reactions overlap under 1 MPa nitrogen pressure, the formation of Bba2 Si2N2O seems to be in competition with that of α-Si3N4.
- At a higher heating rate (100 °C/min), the reaction of formation of Si3N4 and that of Si2N2O clearly overlap, but their exothermic signatures can be easily identified by fitting the exothermic peak with two components. The time sequence of the two reactions is responsible for the main phase formed.
- At the highest heating rates (200 or 300 °C/min), the massive melting of Si occurs, which is characterized by a strong endothermic peak. Only one exothermic peak is observed in these conditions, which is totally or partially superposed with the endothermic peak. The melting of Si and the exothermic reaction start at almost the same time, which suggests that all the reactions take place in the presence of Si in its liquid state.
- The formation of silica inside the preform by partial oxidation of the silicon grains impregnated in the form of slurry lowers the residual porosity of the matrix thanks to the volume expansion induced by the Si oxidation process. In contrast, a silica shell at the surface of the silicon grains limits its reaction with nitrogen, which is responsible for a large amount of residual unreacted silicon left in the formed composite.
- A mixed process that combines the impregnation of a part of silica in the form of slurry and the other by partial oxidation of silicon also introduced into the preform in the form of slurry is a good compromise in terms of Si2N2O content, unreacted silicon left in the matrix and residual open porosity.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Naslain, R. Design, preparation and properties of non-oxide CMCs for application in engines and nuclear reactors: An overview. Compos. Sci. Technol. 2004, 64, 155–170. [Google Scholar] [CrossRef]
- Washburn, M.E. Silicon oxynitride refractories. Am. Ceram. Soc. Bull. 1967, 46, 667. [Google Scholar]
- Lortholary, P.; Billy, M. Etude du Systeme Silicium-Oxygène-Azote. IV. Dégradation Thermique de l’oxynitrure Si2N2O. Bull. Soc. Chim. Fr. 1975, 5–6, 1057–1061. [Google Scholar]
- Billy, M.; Labbe, J.C.; Lortholary, P. Stabilité et Mécanisme de Décomposition Thermique des Oxynitrures de Silicium et de Germanium. Mater. Chem. 1979, 4, 189–200. [Google Scholar] [CrossRef]
- Billy, M.; Boch, P.; Dumazeau, C.; Glandus, J.C.; Goursat, P. Preparation and properties of new silicon oxynitride based ceramics. Ceram. Int. 1981, 7, 13–18. [Google Scholar] [CrossRef]
- Brosset, C.; Idrestedt, I. Crystal structure of silicon oxynitride, Si2N2O. Nature 1964, 201, 1211. [Google Scholar] [CrossRef]
- Sjoberg, J.; Helgesson, G.; Idrestedt, I. Refinement of the structure of Si2N2O. Acta Cryst. 1991, C47, 2438–2441. [Google Scholar] [CrossRef]
- Colquhoun, I.; Wild, S.; Grieveson, P.; Jack, K.H. Thermodynamics of the Silicon-Nitrogen-Oxygen system. In Proceedings of the British Ceramic Society, N°22, Ceramics for High-Temperature Engineering; Godfrey, D.J., Ed.; British Ceramic Society: Stoke-on-Trent, UK, 1973; pp. 207–227. [Google Scholar]
- Weiss, J.; Lukas, H.L.; Lorenz, J.; Petzow, G.; Krieg, P. Calculation of heterogeneous phase equilibria in oxide-nitride systems. I The quaternary system C-Si-N-O. Calphad 1981, 5, 125–140. [Google Scholar] [CrossRef]
- Hillert, M.; Jonsson, S.; Sundman, B. Thermodynamic calculation of the Si-O-N system. Z. Metallkd. 1992, 83, 648–654. [Google Scholar]
- Rocabois, P.; Chatillon, C.; Bernard, C. Thermodynamics of the Si–O–N system: II, stability of Si2N2O(s) by high-temperature mass spectrometric vaporization. J. Am. Ceram. Soc. 1996, 79, 1361–1365. [Google Scholar] [CrossRef]
- Jacobson, N.S. Corrosion of silicon-based ceramics in combustion environments. J. Amer. Ceram. Soc. 1993, 76, 3–28. [Google Scholar] [CrossRef] [Green Version]
- Ogbuji, L.U.J.T.; Bryan, S.R. The SiO2-Si3N4 interface, Part I: Nature of the interphase. J. Am. Ceram. Soc. 1995, 78, 1272–1278. [Google Scholar] [CrossRef]
- Ogbuji, L.U.J.T. The SiO2-Si3N4 interface, Part II: O2 permeation and oxidation reaction. J. Amer. Ceram. Soc. 1995, 78, 1279–1284. [Google Scholar] [CrossRef]
- Moulson, A.J. Review: Reaction-Bonded Silicon Nitride—Its formation and properties. J. Mater. Sci. 1979, 14, 1017–1051. [Google Scholar] [CrossRef]
- Jennings, H.M. Review: On reactions between silicon and nitrogen. J. Mater. Sci. 1983, 18, 951–967. [Google Scholar] [CrossRef]
- Riley, F.L. Silicon Nitride and Related Materials. J. Am. Ceram. Soc. 2000, 83, 245–265. [Google Scholar] [CrossRef]
- Messier, D.R.; Riley, F.L.; Brook, R.J. The α/β silicon nitride phase transformation. J. Mater. Sci. 1978, 13, 1199–1205. [Google Scholar] [CrossRef]
- Sarin, V.K. On the α to ß phase transformation in silicon nitride materials. Sci. Eng. A 1988, 105, 151–159. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, J.R.; Kim, C.H. Effects of Free Silicon on the α to ß Phase Transformation in Silicon Nitride. J. Am. Ceram. Soc. 1987, 70, C240–C242. [Google Scholar] [CrossRef]
- Suematsu, H.; Mitomo, M.; Mitchell, T.E.; Petrovic, J.J.; Fukunaga, O.; Ohashi, N. The α-ß transformation in silicon nitride single crystals. J. Am. Ceram. Soc. 1997, 80, 615–620. [Google Scholar] [CrossRef]
- Ziegler, G.; Heinrich, J.; Wotting, G. Relationships between processing, microstructure and properties of dense and reaction-bonded silicon nitride. J. Mater. Sci. 1987, 22, 3041–3086. [Google Scholar] [CrossRef]
- Rossetti, G.A., Jr.; Denkewicz, R.P., Jr. Kinetic interpretation of α- and β-Si3N4 formation from oxide-free high-purity silicon powder. J. Mater. Sci. 1989, 24, 3081–3086. [Google Scholar] [CrossRef]
- Gemini Sofware Used to Calculate Thermodynamic Equilibrium in Chemical Systems by Minimization of the Gibbs Free Energy, Coach Associated Database Thermodata Grenoble. Available online: http://thermodata.online.fr/gem1ang.html (accessed on 7 July 2021).
- O’Hare, P.A.G.; Tomaszkiewicz, I.; Beck, C.M.; Seifert, H.J. Thermodynamics of silicon nitride. I. Standard molar enthalpies of formation at the temperature 298.15 K of α-Si3N4 and β-Si3N4. J. Chem. Thermodyn. 1999, 31, 303–322. [Google Scholar] [CrossRef]
- Thompson, D.S.; Pratt, P.I. The Structure of Silicon Nitride 33–51 in Science of Ceramics; Stewart, G.H., Ed.; Academic Press: New York, NY, USA, 1967; Volume 3. [Google Scholar]
- Longland, P.; Moulson, A.J. The growth of α- and ß-Si3N4 accompanying the nitriding of silicon powder compacts. J. Mater. Sci. Lett. 1978, 13, 2279–2280. [Google Scholar]
- Atkinson, A.; Moulson, A.J.; Roberts, E.W. Nitriding of high-purity silicon. J. Am. Ceram. Soc. 1976, 59, 285–289. [Google Scholar] [CrossRef]
- Jennings, H.M.; Richman, M.H. Structure, formation mechanisms and kinetics of reaction-bonded silicon nitride. J. Mater. Sci. 1976, 11, 2087–2098. [Google Scholar] [CrossRef]
- Pigeon, R.G.; Varma, A. Quantitative kinetic analysis of silicon nitriding. J. Mater. Sci. 1993, 28, 2999–3013. [Google Scholar] [CrossRef]
- Ekelund, M.; Forslund, B. Kinetics of silicon nitride formation by gas-phase routes in the Si–C–O–N system. J. Mater. Chem. 1992, 2, 1079–1086. [Google Scholar] [CrossRef]
- Sheldon, B.W.; Rankin, J.; Haggerty, J.S. Formation of reaction-bonded silicon nitride from silane derived silicon powders: Nucleation and growth mechanisms. J. Am. Ceram. Soc. 1995, 78, 1624–1632. [Google Scholar] [CrossRef]
- Boyer, S.M.; Moulson, A.J. A mechanism for the nitriding of Fe-contaminated silicon. J. Mater. Sci. 1978, 13, 1637–1646. [Google Scholar] [CrossRef]
- Barinova, T.V.; Barinov, V.Y.; Kovalev, I.D.; Mukhina, N.I. Effect of iron-compound additives on self-propagating high-temperature synthesis of Si2N2O. Refract. Ind. Ceram. 2019, 60, 305–308. [Google Scholar] [CrossRef]
- Dervisbegovic, H.; Riley, F.L. The influence of iron and hydrogen in the nitriding of silicon. J. Mater. Sci. Lett. 1979, 14, 1265–1268. [Google Scholar]
- Dervisbegovic, H.; Riley, F.L. The role of hydrogen in the nitriding of silicon powder compacts. J. Mater. Sci. 1981, 16, 1945–1955. [Google Scholar] [CrossRef]
- Rahaman, M.N.; Moulson, A.J. The removal of surface silica and its effect on the nitridation of high-purity silicon. J. Mater. Sci. 1984, 19, 189–194. [Google Scholar] [CrossRef]
- Messier, D.R.; Wong, P.; Ingram, A.E. Effect of oxygen impurities on the nitriding of high-purity silicon. J. Am. Ceram. Soc. Discuss. Notes 1973, 56, 171–172. [Google Scholar] [CrossRef]
- Wang, M.-J.; Wada, H. Synthesis and characterization of silicon nitride whiskers. J. Mater. Sci. 1990, 25, 1690–1698. [Google Scholar] [CrossRef] [Green Version]
- Mukasyan, A.S.; Borovinskaya, I.P. Structure formation in SHS nitrides. Int. J. SHS 1992, 1, 55–77. [Google Scholar]
- Yao, G.; Li, Y.; Jiang, P.; Jin, X.; Long, M.; Qin, H.; Kumar, R.V. Formation mechanisms of Si3N4 and Si2N2O in silicon powder nitriding. Solid State Sci. 2017, 66, 50–56. [Google Scholar] [CrossRef]
- Ge, C.C.; Li, J.T.; Xia, Y.L. On the mechanism of self-propagating high-temperature synthesis (SHS) of Si3N4. Int. J. Self Propagating High Temp. Synth. 1996, 5, 107–117. [Google Scholar]
- Drevet, B.; Voytovych, R.; Israel, R.; Eustathopoulos, N. Wetting and adhesion of Si on Si3N4 and BN substrates. J. Eur. Ceram. Soc. 2009, 29, 2363–2367. [Google Scholar] [CrossRef]
- Whalen, T.J.; Anderson, A.T. Wetting of SiC, Si3N4, and carbon by Si and Binary Si alloys. J. Am. Ceram. Soc. 1975, 58, 396–399. [Google Scholar] [CrossRef]
- Swartz, J.C. Atmosphere effects on wetting of Si3N4 by Liquid Si. J. Am. Ceram. Soc. Discuss. Notes 1976, 59, 272–273. [Google Scholar] [CrossRef]
- Hirao, K.; Miyamoto, Y.; Koizumi, M. Synthesis of silicon nitride by a combustion reaction under high nitrogen pressure. J. Am. Ceram. Soc. 1986, 69, C60–C61. [Google Scholar]
- Hirao, K.; Miyamoto, Y.; Koizumi, M. Combustion reaction characteristics in the nitriding of silicon. Adv. Ceram. Mater. 1987, 2, 780–784. [Google Scholar] [CrossRef]
- Sangiorgi, R.; Muolo, M.L.; Chatain, D.; Eustathopoulos, N. Wettability and work of adhesion of non-reactive liquid metals on silica. J. Am. Ceram. Soc. 1988, 71, 742–748. [Google Scholar] [CrossRef]
- Weiß, D.; Gebensleben, T.; Diestel, L.; Alphei, L.; Becker, V.; Becker, J.A. The influence of crystallographic orientation on the wetting of silicon on quartz single crystals. J. Mater. Sci. 2011, 46, 3436–3444. [Google Scholar] [CrossRef] [Green Version]
- Wachsmuth, D.; Gebensleben, T.; Weiß, D.; Becker, V.; Alphei, L.D.; Becker, J.A. SiO gas emission and triple line dynamics of small silicon droplets on quartz. J. Cryst. Growth 2012, 355, 122–128. [Google Scholar] [CrossRef]
- Schnurre, S.M.; Gröbner, J.; Schmid-Fetzer, R. Thermodynamics and phase stability in the Si-O system. J. Non Cryst. Solids 2004, 336, 1–25. [Google Scholar] [CrossRef]
- Assael, M.J.; Armyra, I.J.; Brillo, J.; Stankus, S.V.; Wu, J.; Wakeham, W.A. Reference Data for the density and viscosity of liquid Cadmium, Cobalt, Gallium, Indium, Mercury, Silicon, Thallium, and Zinc. J. Phys. Chem. Ref. Data 2012, 41, 033101. [Google Scholar] [CrossRef] [Green Version]
- Narushima, T.; Matsuzawat, K.; Mukaitt, Y.; Lguchi, Y. Oxygen solubility in liquid silicon. Mater. Trans. JIM 1994, 35, 522–528. [Google Scholar] [CrossRef]
- Hirata, H.; Hoshikawa, K. Oxygen solubility and its temperature dependence in a silicon melt in equilibrium with solid silica. J. Cryst. Growth 1990, 106, 657–664. [Google Scholar] [CrossRef]
- Dalaker, H.; Tangstad, M. Temperature dependence of the solubility of nitrogen in liquid silicon equilibrated with silicon nitride. Mater. Trans. 2009, 50, 2541–2544. [Google Scholar] [CrossRef]
- Pradeilles, N.; Record, M.C.; Marin-Ayral, R.M. A modified SHS method for Si2N2O elaboration. J. Eur. Ceram. Soc. 2006, 26, 2489–2495. [Google Scholar] [CrossRef]
- Miyazaki, H.; Yoshida, S.; Sato, Y.; Suzuki, H.; Ota, T. Fabrication of radiative cooling materials based on Si2N2O particles by the nitriding of mixtures of silicon and silicon dioxide powders. J. Ceram. Soc. Jpn. 2013, 121, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Radwan, M.; Kashiwagi, T.; Miyamoto, Y. New synthesis route for Si2N2O ceramics based on desert sand. J. Eur. Ceram. Soc. 2003, 23, 2337–2341. [Google Scholar] [CrossRef]
- Persson, J.; Käll, P.O.; Nygren, M. Interpretation of the parabolic and nonparabolic oxidation behavior of silicon oxynitride. J. Am. Ceram. Soc. 1992, 75, 3377–3384. [Google Scholar] [CrossRef]
- Du, H.; Tressler, R.E.; Spear, K.E.; Pantano, C.G. Oxidation studies of crystalline CVD nitride. J. Electrochem. Soc. 1989, 136, 1527–1536. [Google Scholar] [CrossRef]
- Bergman, B.; Heping, H. The influence of different oxides on the formation of Si2N2O from SiO2 and Si3N4. J. Eur. Ceram. Soc. 1990, 6, 3–8. [Google Scholar] [CrossRef]
- Deng, S.; He, G.; Wang, L.; Meng, Q.; Yang, Z.; Li, J.; Jiang, L. Combustion synthesis of Si2N2O powder for photovoltaic silicon casting applicatio. Ceram. Int. 2021, 47, 5795–5802. [Google Scholar] [CrossRef]
- Batha, H.D.; Whitney, E.D. Kinetics and mechanism of the thermal decomposition of Si3N4. J. Am. Ceram. Soc. 1973, 56, 365–369. [Google Scholar] [CrossRef]
- Li, X.; Zhang, L.; Yin, X. Study on in-situ reaction synthesis and mechanical properties of Si2N2O ceramic. Ceram. Int. 2012, 39, 3035–3041. [Google Scholar] [CrossRef]
- Hackley, V.A.; Paik, U.; Kim, B.H.; Malghan, S.G. Aqueous processing of sintering reaction-bonded silicon nitride: Dispersion properties of silicon powder. J. Am. Ceram. Soc. 1997, 80, 1781–1788. [Google Scholar] [CrossRef]
- Yao, D.X.; Xia, Y.F.; Zeng, Y.P.; Zuo, K.H.; Jiang, D.L. Porous Si3N4 ceramics prepared via slip casting of Si and reaction bonded silicon nitride. Ceram. Int. 2011, 37, 3071–3076. [Google Scholar] [CrossRef]
- Xu, G.L.; Zhang, J.J.; Song, G.Z. Effect of complexation on the zeta potential of silica powder. Powder Technol. 2003, 134, 218–222. [Google Scholar] [CrossRef]
- Tourbin, M. Caractérisation et Comportement de Suspensions Concentrées de Nanoparticules sous Écoulement: Application aux Processus D’agrégation et de Rupture. Ph.D. Thesis, Université de Toulouse, Toulouse, France, 2006. [Google Scholar]
- Ramachandra, R.R.; Roopa, H.N.; Kannan, T.S. The characterisation of aqueous silicon slips. J. Eur. Ceram. Soc. 1999, 19, 2763–2771. [Google Scholar]
- Bowden, M.E.; Barris, G.C.; Brown, W.M.; Jefferson, D.A. A new, low-temperature polymorph of O’-SiAlON. J. Am. Ceram. Soc. 1998, 81, 2188–2190. [Google Scholar] [CrossRef]
- Goursat, P.; Lortholary, P.; Billy, M. Etude du système silicium-oxygène-azote I. Préparation de l’oxynitrure Si2N2O. Rev. Int. Tempér. Réfract. 1971, 8, 149–154. [Google Scholar]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heating Rate (°C/min) | Time Required to Reach the Melting Temperature of Si (1414 °C) (s) | Time Required to Reach the Step Temperature of 1430 °C (s) |
|---|---|---|
| 50 | 377 | 396 |
| 100 | 188 | 198 |
| 200 | 94 | 99 |
| 300 | 63 | 66 |
| GB 1 | GB 2 | GB 3 | Pref. 1 | Pref. 2 | Pref. 3 | |
|---|---|---|---|---|---|---|
| Weight before oxidation (g) | 2.62 | 1.55 | 0.95 | 2.53 | 2.61 | 2.59 |
| Weight after oxidation (g) | 3.02 | 1.80 | 1.10 | 2.69 | 2.78 | 2.75 |
| Total weight gain (%) | 15.4 | 16.4 | 15.8 | 17.6 | 17.3 | 17.2 |
| Initial mass of silicon (g) | 1.91 | 1.13 | 0.69 | 0.64 | 0.69 | 0.68 |
| Silicon weight gain (%) | 21.2 | 22.5 | 21.7 | 24.2 | 23.8 | 23.6 |
| Si2N2O (Natural Sinoite) Brosset and Idrestedt (1964) [6] | Si2N2O (Synthetic Sinoite) Sjoberg et al. (1991) [7] | Si1.8Al0.2O1.2N1.8 Bowden et al. (1998) [70] | Si2N2O Our Simulation | |
|---|---|---|---|---|
| Space group | Cmc21 | Cmc21 | Bba2 | Bba2 |
| a (nm) | 0.8843 | 0.88717 | 0.88488 | 0.88102 |
| b (nm) | 0.5473 | 0.54909 | 0.53752 | 0.53698 |
| c (nm) | 0.48435 | 0.48504 | 0.48355 | 0.48355 |
| N° Sample | Heating Rate (°C/min) | N2 Pressure (MPa) | Si2N2O Total | Si3N4 Total | Si wt% | Residual Open Porosity (%) | Filled Open Porosity (%) | Si2N2O Cmc21 wt% | Si2N2O Bba2 wt% | Si2N2O Bba2/ Cmc21 | Si3N4 Alpha wt% | Si3N4 Beta wt% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 50 | 1 | 41.3 | 58.7 | 0.0 | 30.7 | 41 | 10.1 | 31.2 | 3.1 | 51.8 | 6.9 |
| 2 | 100 | 1 | 74.8 | 24.8 | 0.4 | 27.3 | 48 | 33.7 | 41.1 | 1.2 | 17.3 | 7.5 |
| 3 | 200 | 1 | 87.4 | 9.4 | 3.2 | 29.2 | 44 | 36.3 | 51.1 | 1.4 | 6.2 | 3.2 |
| 4 | 300 | 1 | 79.1 | 17.7 | 3.2 | 29.9 | 42 | 36.3 | 42.8 | 1.2 | 12.5 | 5.2 |
| 5 | 50 | 2 | 17.8 | 82.1 | 0.1 | 29.7 | 43 | 7.7 | 10.1 | 1.3 | 71.8 | 10.3 |
| 6 | 100 | 2 | 67.7 | 32.2 | 0.1 | 27.2 | 48 | 18.0 | 49.7 | 2.8 | 25.3 | 6.9 |
| 7 | 200 | 2 | 93.4 | 6.4 | 0.2 | 24.4 | 53 | 33.3 | 60.1 | 1.8 | 2.7 | 3.7 |
| 8 | 300 | 2 | 94.7 | 4.9 | 0.4 | 30.0 | 42 | 30.2 | 64.5 | 2.1 | 2.6 | 2.3 |
| 9 | 50 | 3 | 16.4 | 83.3 | 0.3 | 29.1 | 44 | 7.6 | 8.8 | 1.2 | 71.8 | 11.5 |
| 10 | 100 | 3 | 38.3 | 61.7 | 0.0 | 26.9 | 48 | 5.2 | 33.1 | 6.4 | 52.6 | 9.1 |
| 11 | 200 | 3 | 80.9 | 18.6 | 0.5 | 27.9 | 46 | 26.1 | 54.8 | 2.1 | 14.3 | 4.3 |
| 12 | 300 | 3 | 86.6 | 12.5 | 0.9 | 29.6 | 43 | 24.9 | 61.7 | 2.5 | 7.2 | 5.3 |
| N° Sample | Heating Rate (°C/min) | N2 Pressure (MPa) | Area (A.U.) | Center (s) | Width (s) | Height (°C) |
|---|---|---|---|---|---|---|
| Peak 1 | ||||||
| 1 | 50 | 1 | 3036 | 223 | 75 | 26 |
| 5 | 50 | 2 | 2119 | 185 | 56 | 24 |
| 9 | 50 | 3 | 1342 | 169 | 54 | 16 |
| Peak 2 | ||||||
| 1 | 50 | 1 | 1858 | 311 | 28 | 42 |
| 5 | 50 | 2 | 1772 | 304 | 21 | 53 |
| 9 | 50 | 3 | 1883 | 292 | 16 | 75 |
| N° Sample | Process | Heating Rate (°C/min) | Si2N2O Total | Si3N4 Total | Si wt% | Residual Open Porosity (%) | Filled Open Porosity (%) | Si2N2O Cmc21 wt% | Si2N2O Bba2 wt% | Si2N2O Bba2/Cmc21 | Si3N4 Alpha wt% | Si3N4 Beta wt% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | STD | 300 | 94.7 | 4.9 | 0.4 | 30 | 42 | 30.2 | 64.5 | 2.1 | 2.6 | 2.3 |
| 13 | MP1 | 300 | 65.8 | 9.9 | 24.3 | 13.5 | 73.9 | 20 | 45.8 | 2.3 | 9.9 | 0 |
| 14 | MP2 | 300 | 84.4 | 13.9 | 1.7 | 23.8 | 54.2 | 38.4 | 46 | 1.2 | 5.4 | 8.5 |
| 15 | MP2 | 200 | 87.5 | 10.2 | 2.3 | 22.9 | 55.8 | 39.8 | 47.7 | 1.2 | 4.7 | 5.5 |
| 16 | MP2 | 100 | 81.6 | 17.4 | 1 | 24.7 | 52.5 | 39.3 | 42.3 | 1.1 | 14.3 | 3.1 |
| 17 | MP2 | 50 | 55.9 | 43.3 | 0.8 | 21 | 59.5 | 23.2 | 32.7 | 1.4 | 37.9 | 5.4 |
| N° | Process | Heating Rate (°C/min) | Si2N2O Total | Si3N4 Total | Si wt% | Residual Open Porosity (%) | Filled Open Porosity (%) | Si2N2O Cmc21 wt% | Si2N2O Bba2 wt% | Si2N2O Bba2/ Cmc21 | Si3N4 Alpha wt% | Si3N4 Beta wt% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | STD | 200 | 93.4 | 6.4 | 0.2 | 24.4 | 53 | 33.3 | 60.1 | 1.8 | 2.7 | 3.7 |
| 18 | STD | 200 | 78.2 | 21.2 | 0.6 | 15.9 | 54.6 | 23.1 | 55.1 | 2.4 | 16.3 | 4.9 |
| 19 | MP2 | 200 | 72.3 | 17.5 | 10.2 | 9.9 | 71.6 | 40.7 | 31.6 | 0.8 | 12.2 | 5.3 |
| 8 | STD | 300 | 94.7 | 4.9 | 0.4 | 30.0 | 42 | 30.2 | 64.5 | 2.1 | 2.6 | 2.3 |
| 20 | STD | 300 | 68.5 | 28.5 | 3.0 | 14.3 | 59.2 | 23.8 | 44.7 | 1.9 | 19.3 | 9.2 |
| 21 | MP2 | 300 | 80.1 | 11.5 | 8.4 | 10.4 | 70.2 | 37.6 | 42.5 | 1.1 | 6.9 | 4.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taillet, B.; Pailler, R.; Teyssandier, F. Densification of Ceramic Matrix Composite Preforms by Si2N2O Formed by Reaction of Si with SiO2 under High Nitrogen Pressure. Part 1: Materials Synthesis. J. Compos. Sci. 2021, 5, 178. https://doi.org/10.3390/jcs5070178
Taillet B, Pailler R, Teyssandier F. Densification of Ceramic Matrix Composite Preforms by Si2N2O Formed by Reaction of Si with SiO2 under High Nitrogen Pressure. Part 1: Materials Synthesis. Journal of Composites Science. 2021; 5(7):178. https://doi.org/10.3390/jcs5070178
Chicago/Turabian StyleTaillet, Brice, René Pailler, and Francis Teyssandier. 2021. "Densification of Ceramic Matrix Composite Preforms by Si2N2O Formed by Reaction of Si with SiO2 under High Nitrogen Pressure. Part 1: Materials Synthesis" Journal of Composites Science 5, no. 7: 178. https://doi.org/10.3390/jcs5070178
APA StyleTaillet, B., Pailler, R., & Teyssandier, F. (2021). Densification of Ceramic Matrix Composite Preforms by Si2N2O Formed by Reaction of Si with SiO2 under High Nitrogen Pressure. Part 1: Materials Synthesis. Journal of Composites Science, 5(7), 178. https://doi.org/10.3390/jcs5070178