
A pH-Responsive Foam Formulated with PAA/Gemini 12-2-12 Complexes



Abstract
:1. Introduction
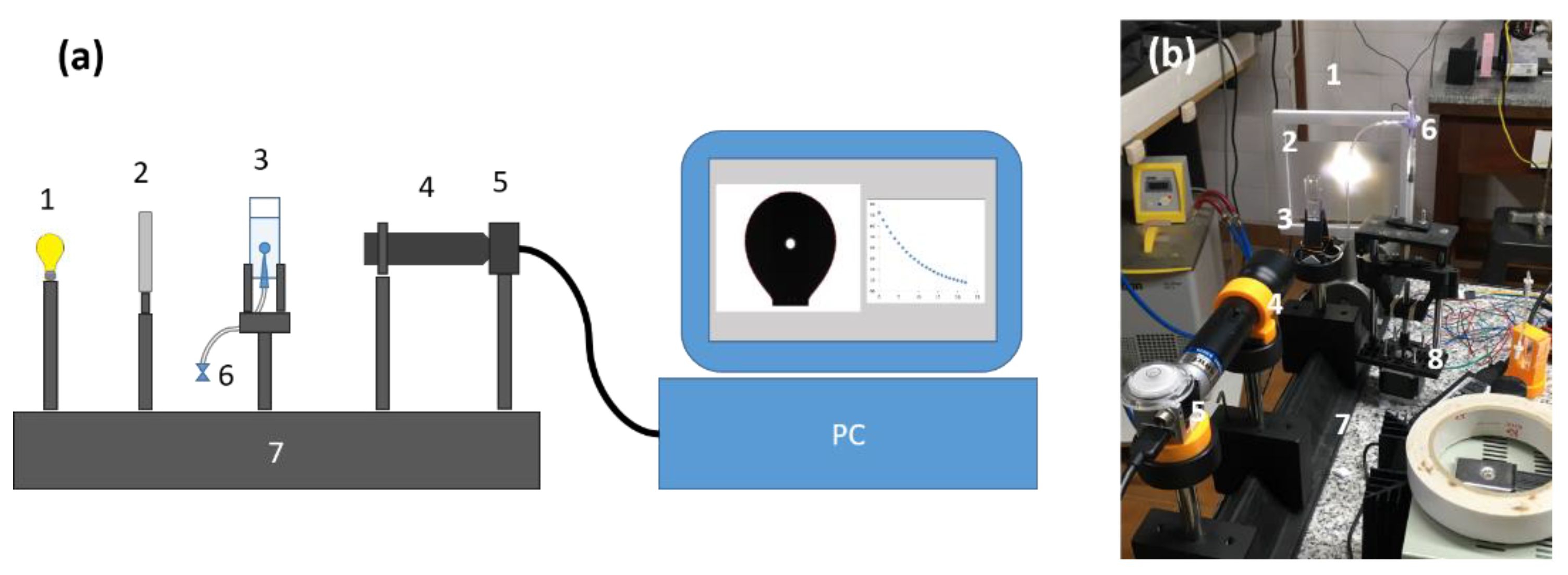
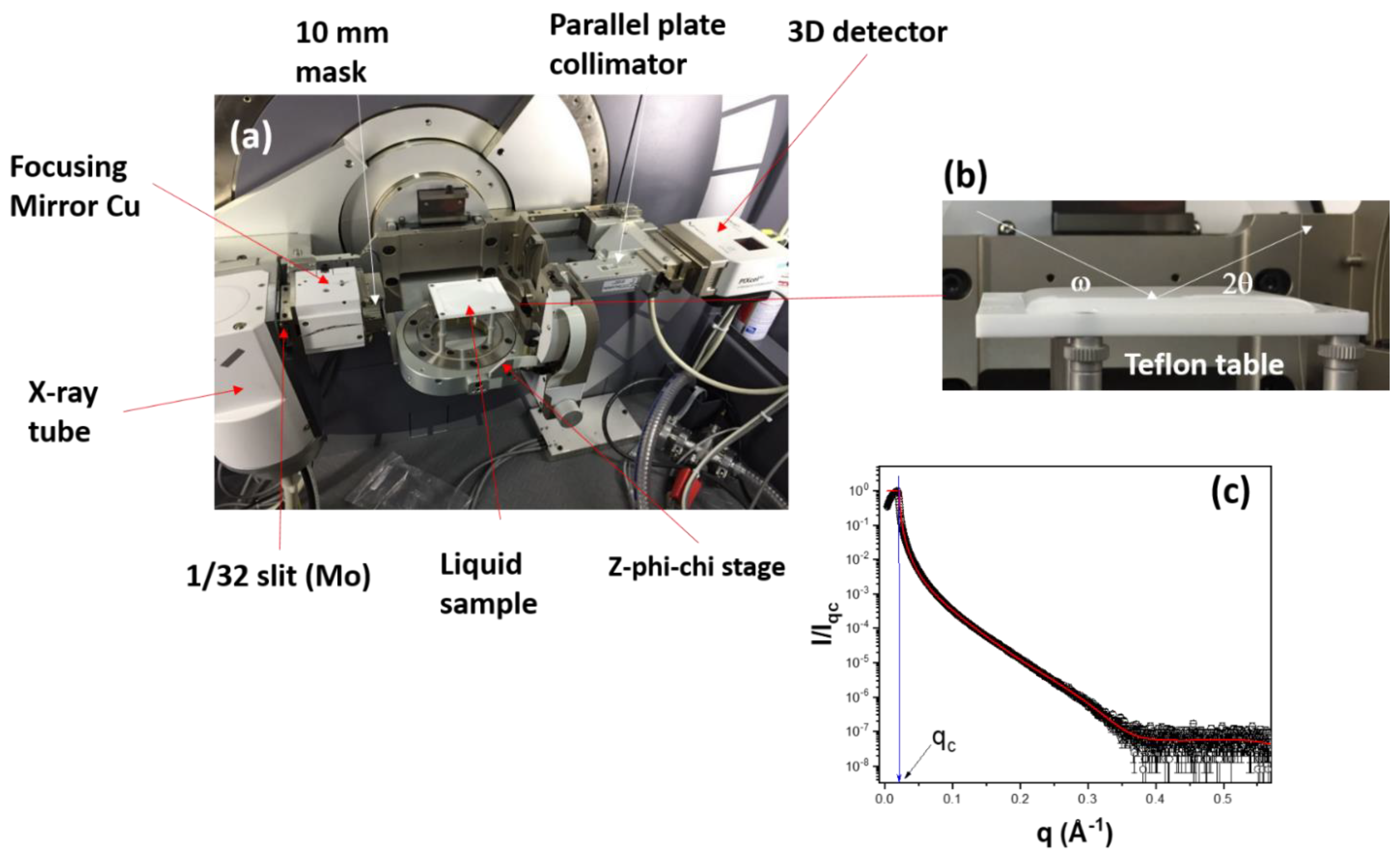
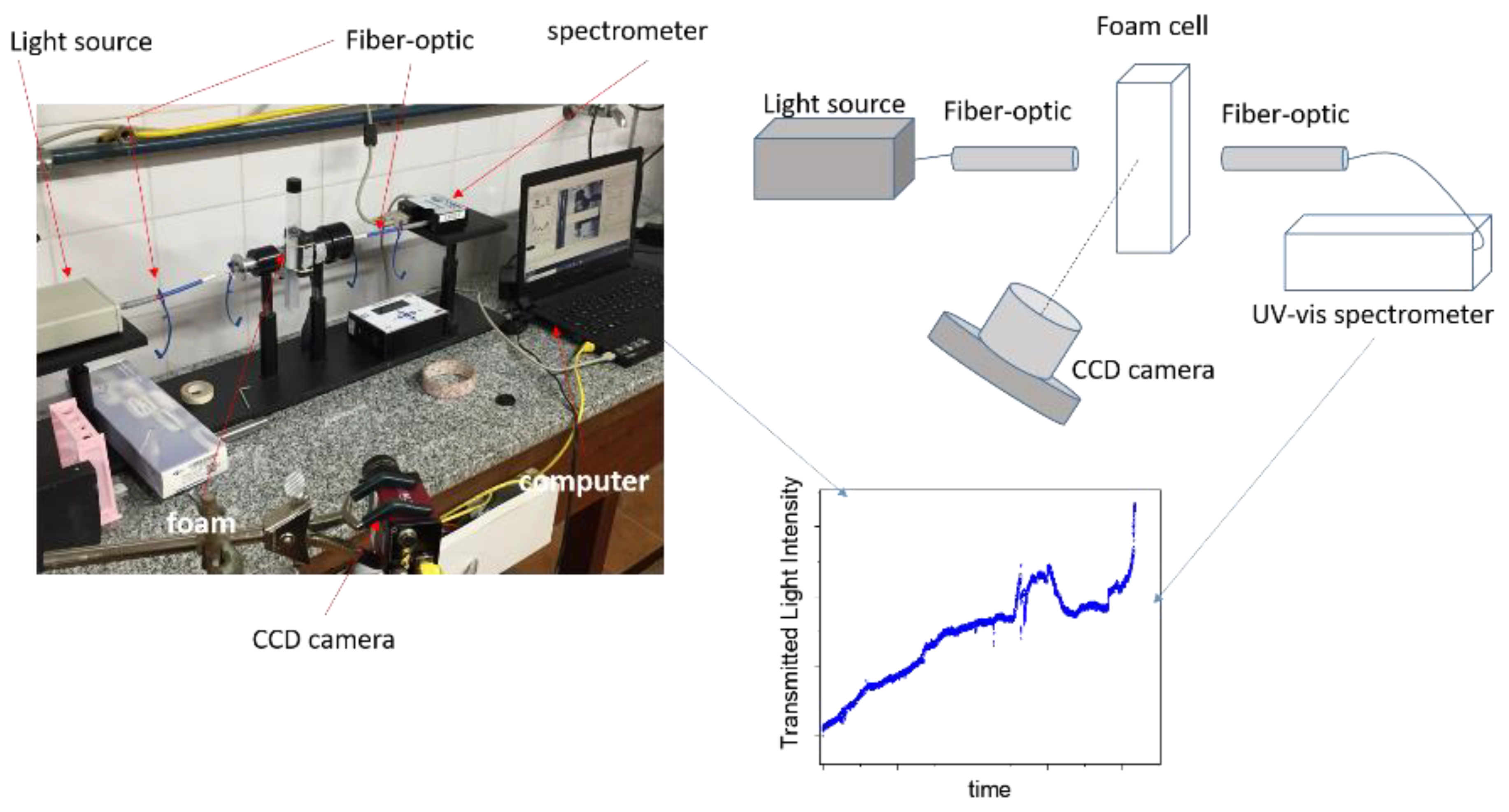
2. Materials and Methods
3. Results
3.1. Phase Behavior
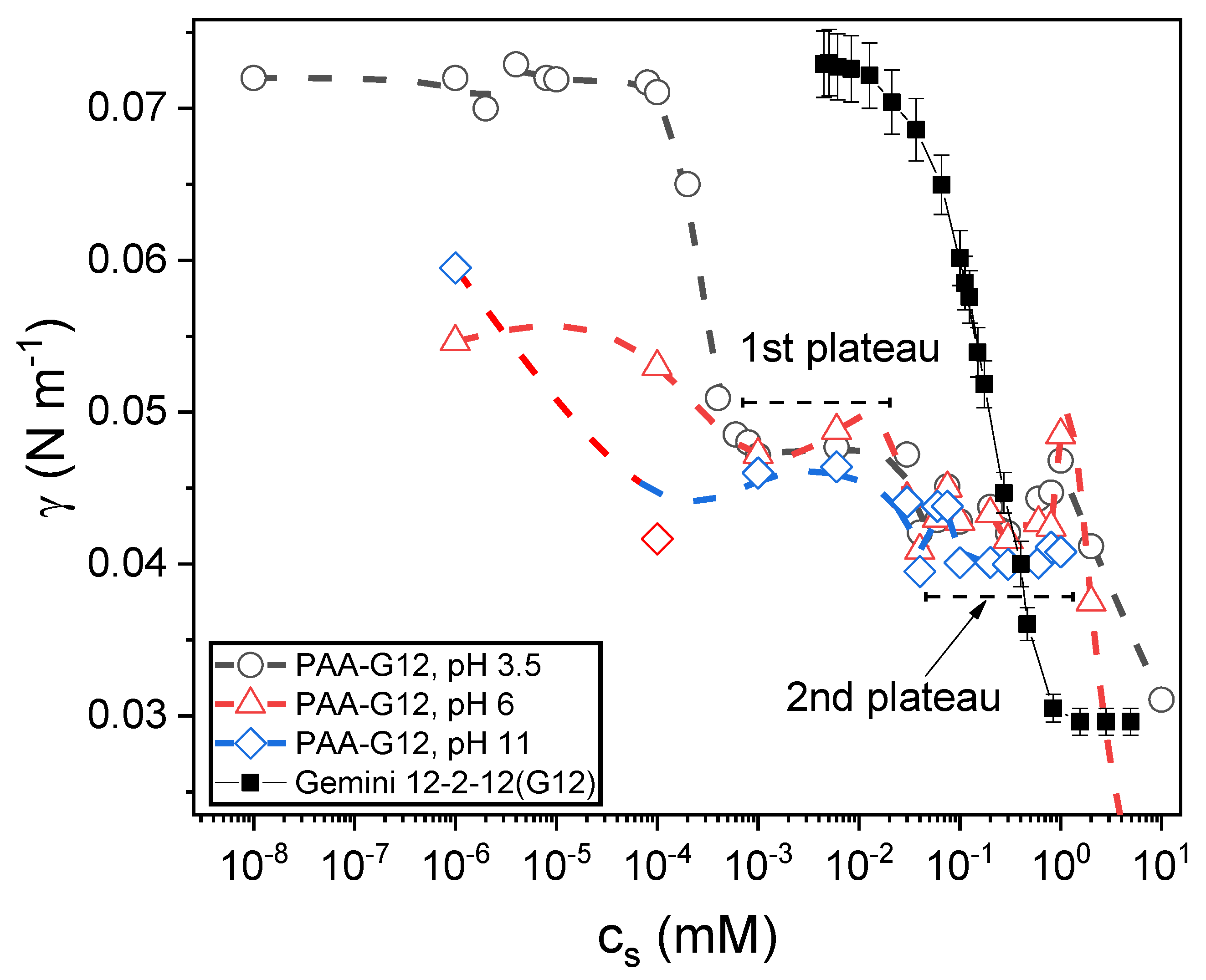
3.2. Equilibrium Surface Tension
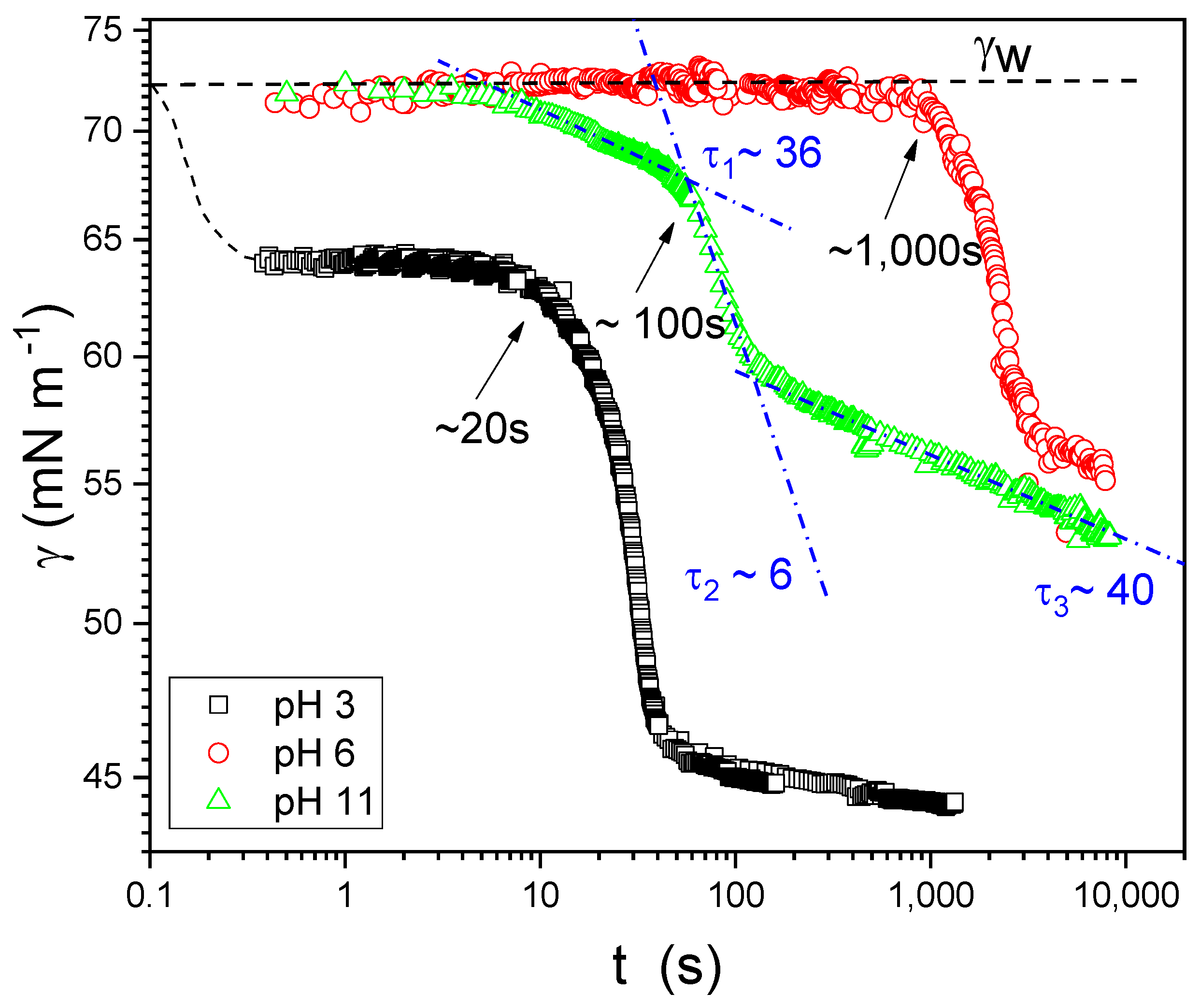
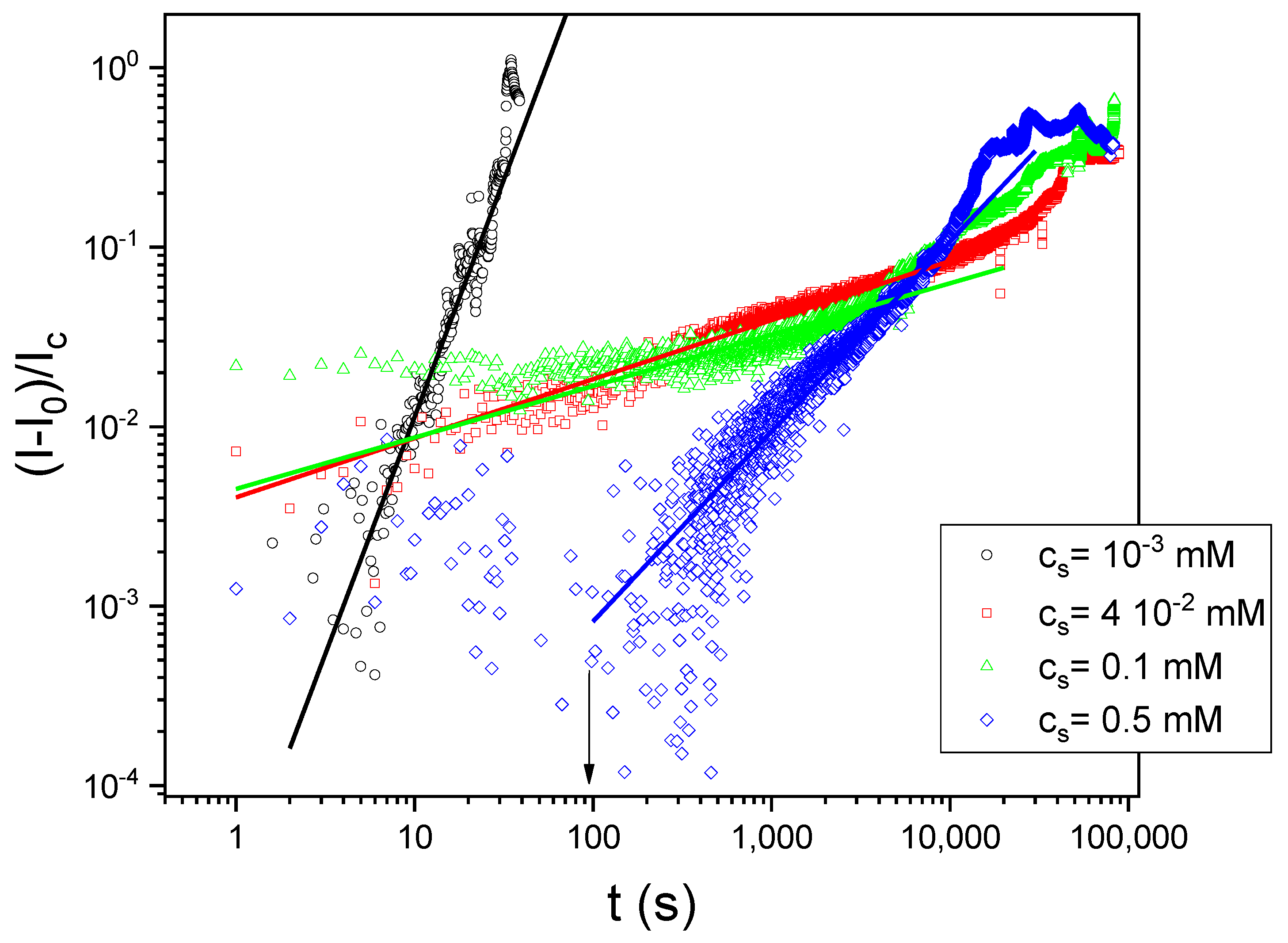
3.3. Dynamic Surface Tension
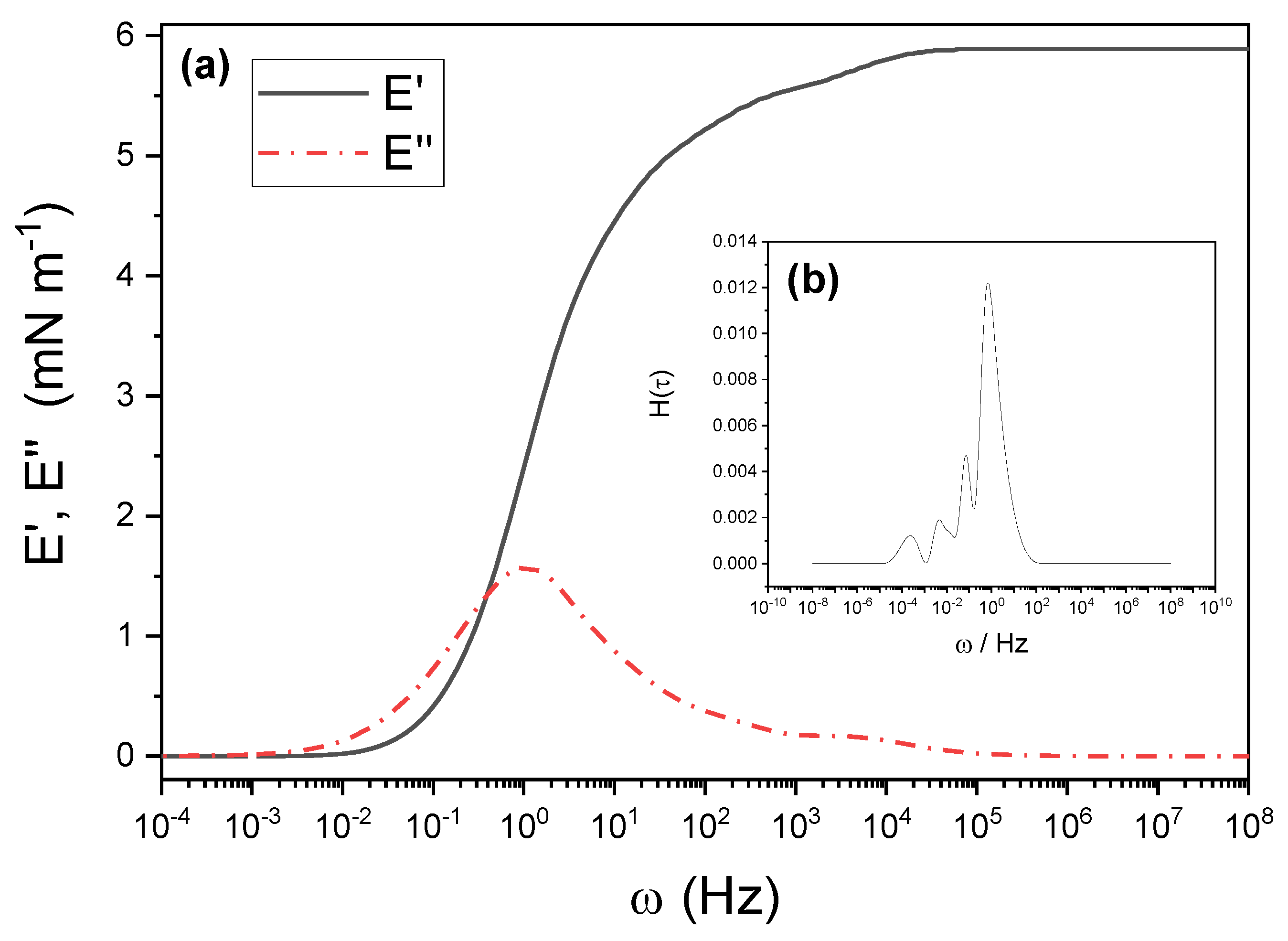
3.4. Surface Viscoelasticity
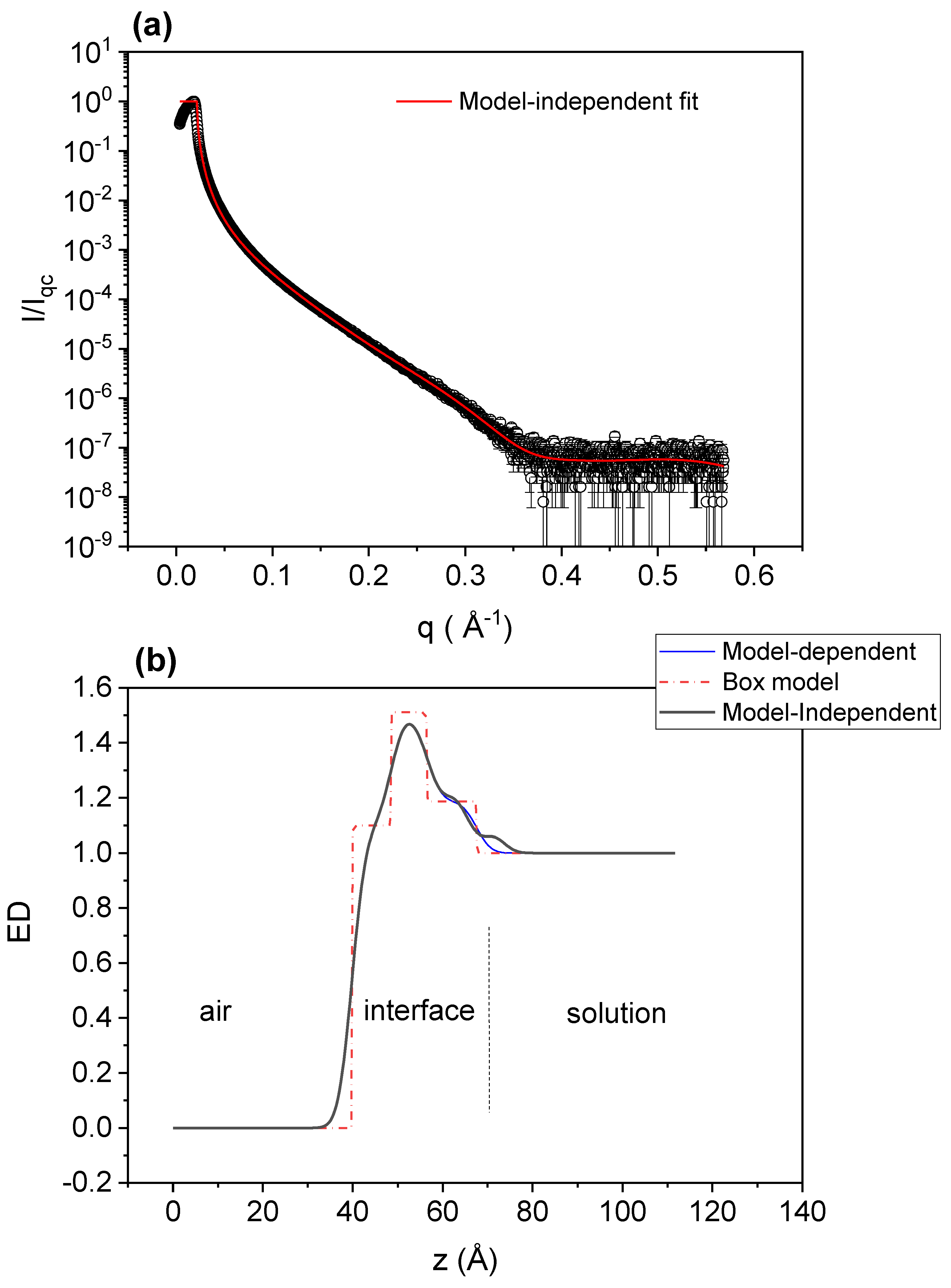
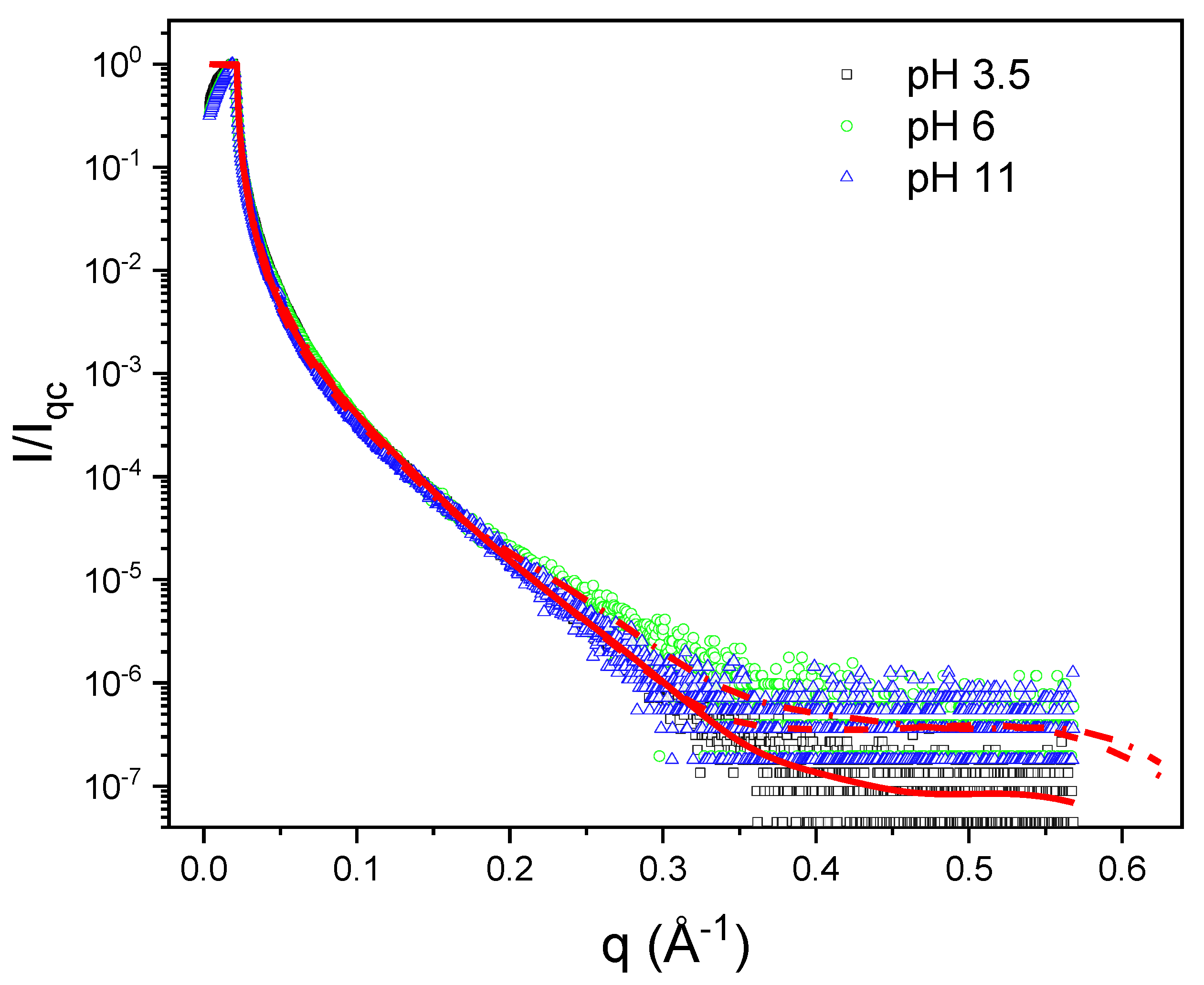
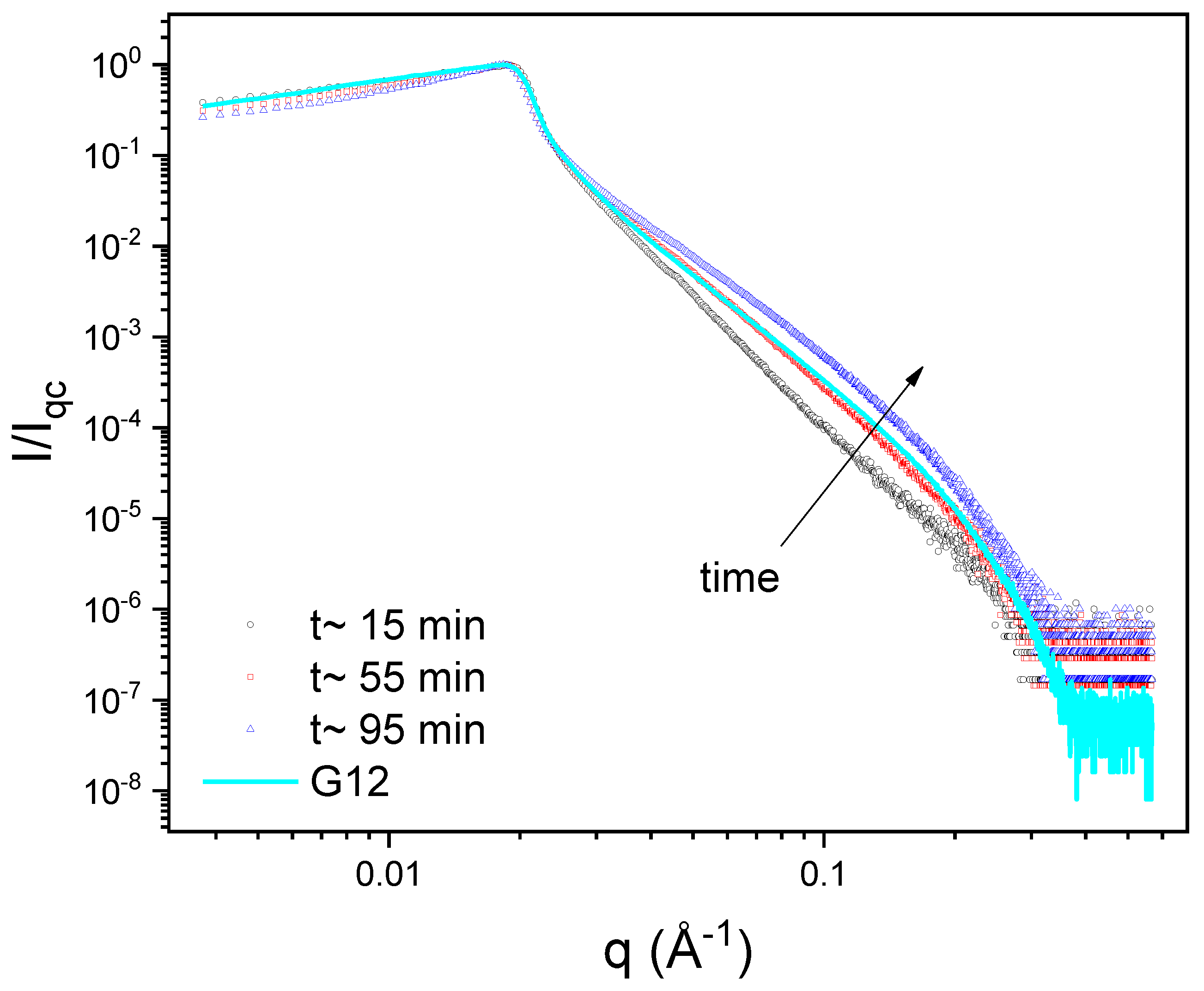
3.5. X-ray Reflectometry (XRR)
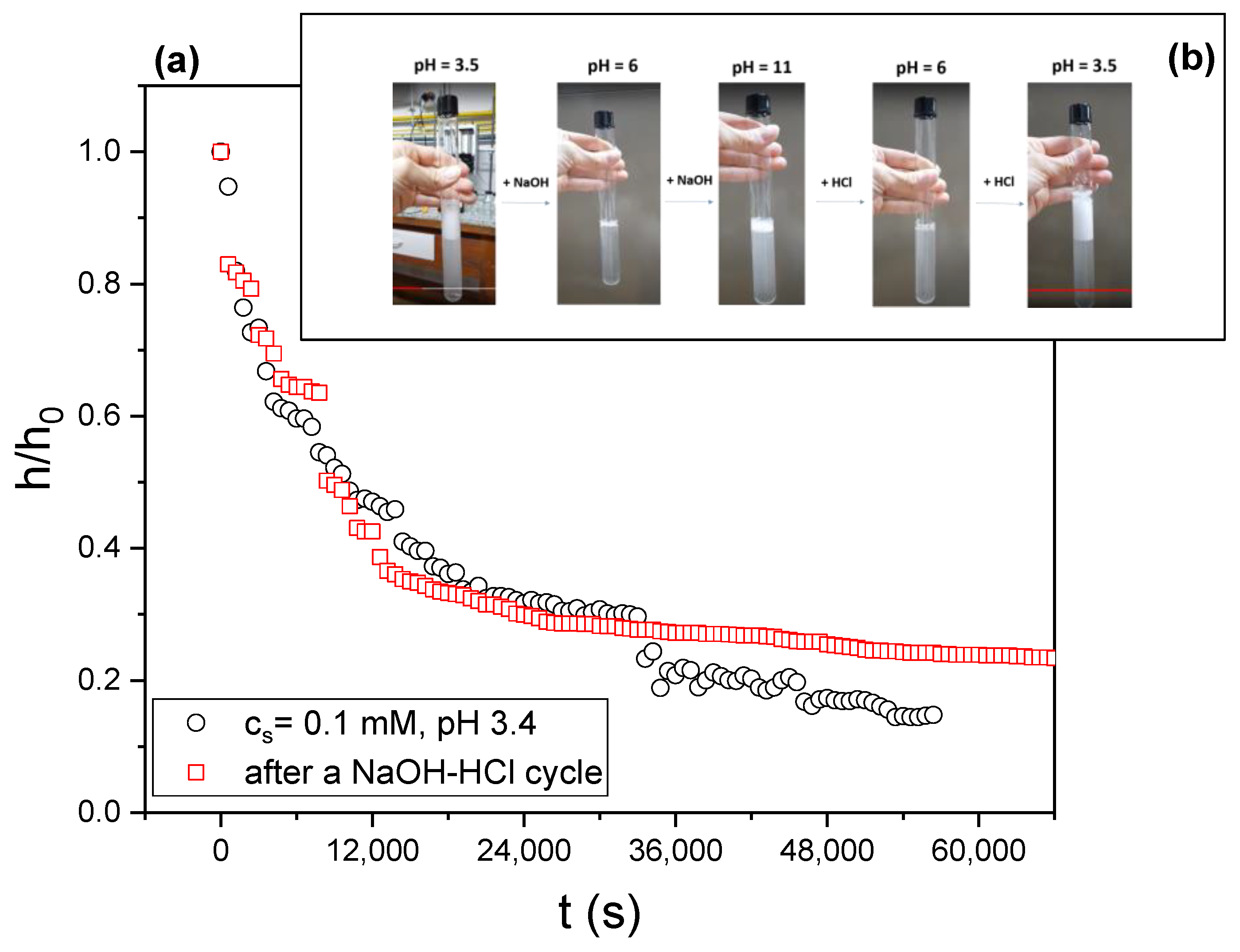
3.6. Foam Stability
4. Discussion
5. Conclusions and Outlooks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosen, M.J. Surfactants and Interfacial Phenomena, 3rd ed.; Wiley-Interscience: Hoboken, NJ, USA, 2004; ISBN 0471478180. [Google Scholar]
- Ritacco, H.A. Electro-optic Kerr effect in the study of mixtures of oppositely charged colloids. The case of polymer-surfactant mixtures in aqueous solutions. Adv. Colloid Interface Sci. 2017, 247, 234–257. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.; Llamas, S.; Maestro, A.; Fernández-Peña, L.; Akanno, A.; Miller, R.; Ortega, F.; Rubio, R.G. Polymer–surfactant systems in bulk and at fluid interfaces. Adv. Colloid Interface Sci. 2016, 233, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Piculell, L. Understanding and Exploiting the Phase Behavior of Mixtures of Oppositely Charged Polymers and Surfactants in Water. Langmuir 2013, 29, 10313–10329. [Google Scholar] [CrossRef]
- Kristen-Hochrein, N.; Laschewsky, A.; Miller, R.; Von Klitzing, R. Stability of Foam Films of Oppositely Charged Polyelectrolyte/Surfactant Mixtures: Effect of Isoelectric Point. J. Phys. Chem. B 2011, 115, 14475–14483. [Google Scholar] [CrossRef]
- Bain, C.D.D.; Claesson, P.M.M.; Langevin, D.; Meszaros, R.; Nylander, T.; Stubenrauch, C.; Titmuss, S.; von Klitzing, R. Complexes of surfactants with oppositely charged polymers at surfaces and in bulk. Adv. Colloid Interface Sci. 2010, 155, 32–49. [Google Scholar] [CrossRef]
- Gradzielski, M.; Hoffmann, I. Polyelectrolyte-surfactant complexes (PESCs) composed of oppositely charged components. Curr. Opin. Colloid Interface Sci. 2018, 35, 124–141. [Google Scholar] [CrossRef]
- Goddard, E.D.; Ananthapadmanabhan, K.P. Interactions of Surfactants with Polymers and Proteins; CRC Press: Boca Raton, FL, USA, 1993; ISBN 978-0849367847. [Google Scholar]
- Kwak, J.C.T.; Jan, C.T. (Eds.) Polymer-Surfactant Systems. Surfactant Science Series Vol. 77; M. Dekker: New York, NY, USA, 1998; ISBN 9780824702328. [Google Scholar]
- Langevin, D. Complexation of oppositely charged polyelectrolytes and surfactants in aqueous solutions. A review. Adv. Colloid Interface Sci. 2009, 147–148, 170–177. [Google Scholar] [CrossRef]
- Lencina, M.M.M.S.S.; Fernández Miconi, E.; Fernández Leyes, M.D.; Domínguez, C.; Cuenca, E.; Ritacco, H.A. Effect of surfactant concentration on the responsiveness of a thermoresponsive copolymer/surfactant mixture with potential application on “Smart” foams formulations. J. Colloid Interface Sci. 2018, 512, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Nakamura, Y. Stimuli-Responsive Bubbles and Foams Stabilized with Solid Particles. Langmuir 2017, 33, 7365–7379. [Google Scholar] [CrossRef]
- Fameau, A.-L.; Carl, A.; Saint-Jalmes, A.; von Klitzing, R. Responsive Aqueous Foams. ChemPhysChem 2015, 16, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Fameau, A.L.; Fujii, S. Stimuli-responsive liquid foams: From design to applications. Curr. Opin. Colloid Interface Sci. 2020. [Google Scholar] [CrossRef]
- Huang, J.; Cheng, F.; Binks, B.P.; Yang, H. pH-Responsive Gas–Water–Solid Interface for Multiphase Catalysis. J. Am. Chem. Soc. 2015, 137, 15015–15025. [Google Scholar] [CrossRef]
- Tang, C.; Xiao, E.; Sinko, P.J.; Szekely, Z.; Prud’homme, R.K. Responsive foams for nanoparticle delivery. Colloids Surfaces B Biointerfaces 2015, 133, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Seekell, R.P.; Cole, A.R.; Lamothe, J.R.; Lock, A.T.; van den Bosch, S.; Tang, X.; Kheir, J.N.; Polizzotti, B.D. Interfacial Nanoprecipitation toward Stable and Responsive Microbubbles and Their Use as a Resuscitative Fluid. Angew. Chem. Int. Ed. 2018, 57, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Langevin, D. Aqueous Foams: A Field of Investigation at the Frontier between Chemistry and Physics. ChemPhysChem 2008, 9, 510–522. [Google Scholar] [CrossRef]
- Weaire, D.L.; Hutzler, S. The Physics of Foams; Clarendon Press: Oxford, UK, 1999; ISBN 0198505515. [Google Scholar]
- Exerowa, D.R.; Kruglyakov, P.M. Foam and Foam Films: Theory, Experiment, Application, 1st ed.; Möbius, D., Miller, R., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 1998; Volume 5, ISBN 978-0-444-81922-2. [Google Scholar]
- Hill, C.; Eastoe, J. Foams: From nature to industry. Adv. Colloid Interface Sci. 2017, 247, 496–513. [Google Scholar] [CrossRef] [Green Version]
- Saint-Jalmes, A. Physical chemistry in foam drainage and coarsening. Soft Matter 2006, 2, 836. [Google Scholar] [CrossRef] [PubMed]
- Langevin, D. Bubble coalescence in pure liquids and in surfactant solutions. Curr. Opin. Colloid Interface Sci. 2015, 20, 92–97. [Google Scholar] [CrossRef]
- Bureiko, A.; Trybala, A.; Kovalchuk, N.; Starov, V. Current applications of foams formed from mixed surfactant-polymer solutions. Adv. Colloid Interface Sci. 2015, 222, 670–677. [Google Scholar] [CrossRef]
- Langevin, D. Influence of interfacial rheology on foam and emulsion properties. Adv. Colloid Interface Sci. 2000, 88, 209–222. [Google Scholar] [CrossRef]
- Binks, B.P.; Murakami, R.; Armes, S.P.; Fujii, S.; Schmid, A.; Binks, B.P.; Murakami, R.; Armes, S.P.; Fujii, S.; Schmid, A.; et al. PH-responsive aqueous foams stabilized by ionizable latex particles. Langmuir 2007, 23, 8691–8694. [Google Scholar] [CrossRef]
- Cuenca, V.E.; Falcone, R.D.; Silber, J.J.; Correa, N.M. How the Type of Cosurfactant Impacts Strongly on the Size and Interfacial Composition in Gemini 12-2-12 RMs Explored by DLS, SLS, and FTIR Techniques. J. Phys. Chem. B 2016, 120, 467–476. [Google Scholar] [CrossRef]
- Zana, R.; Benrraou, M.; Rueff, R. Alkanediyl-.alpha.,.omega.-bis(dimethylalkylammonium bromide) surfactants. 1. Effect of the spacer chain length on the critical micelle concentration and micelle ionization degree. Langmuir 1991, 7, 1072–1075. [Google Scholar] [CrossRef]
- Cuenca, V.E.; Fernández-Leyes, M.; Falcone, R.D.D.D.; Correa, N.M.; Langevin, D.; Ritacco, H.H.A.; Fernández Leyes, M.; Falcone, R.D.D.D.; Correa, N.M.; Langevin, D.; et al. Interfacial dynamics and its relations with “negative” surface viscosities measured at water-air interfaces covered with a cationic surfactant. Langmuir 2019, 35, 8333–8343. [Google Scholar] [CrossRef]
- Daerr, A.; Mogne, A. Pendent_Drop: An ImageJ Plugin to Measure the Surface Tension from an Image of a Pendent Drop. J. Open Res. Softw. 2016, 4, 2–6. [Google Scholar]
- Monroy, F.; Ortega, F.; Rubio, R.G. Dilatational rheology of insoluble polymer monolayers: Poly(vinylacetate). Phys. Rev. E 1998, 58, 7629–7641. [Google Scholar] [CrossRef] [Green Version]
- Provencher, S.W. A constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput. Phys. Commun. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Provencher, S.W. Contin: A general purpose constrained regularization program for inverting noisy linear algebraic and integral equations. Comput. Phys. Commun. 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Danauskas, S.M.; Li, D.; Meron, M.; Lin, B.; Lee, K.Y.C. Stochastic fitting of specular X-ray reflectivity data using StochFit. J. Appl. Crystallogr. 2008, 41, 1187–1193. [Google Scholar] [CrossRef]
- Durian, D.J.; Weitz, D.A.; Pine, D.J. Scaling behavior in shaving cream. Phys. Rev. A 1991, 44, R7902–R7905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, M.U.; Saint-Jalmes, A.; Durian, D.J. Scattering optics of foam. Appl. Opt. 2001, 40, 4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritacco, H. Playing with Liquid Foams: Learning Physical Chemistry. J. Chem. Educ. 2008, 85, 1667. [Google Scholar] [CrossRef]
- Georgieva, D.; Cagna, A.; Langevin, D. Link between surface elasticity and foam stability. Soft Matter 2009, 5, 2063. [Google Scholar] [CrossRef]
- Georgieva, D.; Schmitt, V.V.; Leal-Calderon, F.; Langevin, D. On the possible role of surface elasticity in emulsion stability. Langmuir 2009, 25, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Langevin, D. Polyelectrolyte and surfactant mixed solutions. Behavior at surfaces and in thin films. Adv. Colloid Interface Sci. 2001, 89–90, 467–484. [Google Scholar] [CrossRef]
- Ritacco, H.; Albouy, P.-A.; Bhattacharyya, A.; Langevin, D. Influence of the polymer backbone rigidity on polyelectrolyte–surfactant complexes at the air/water interface. Phys. Chem. Chem. Phys. 2000, 2, 5243–5251. [Google Scholar] [CrossRef]
- Stubenrauch, C.; Albouy, P.-A.A.; Klitzing, R.V.; Langevin, D. Polymer/surfactant complexes at the water/air interface: A surface tension and X-ray reflectivity study. Langmuir 2000, 16, 3206–3213. [Google Scholar] [CrossRef]
- Tanford, C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes, 2nd ed.; Wiley Interscience Publication; Wiley: Somerset, NJ, USA, 1980; ISBN 978-0471048930. [Google Scholar]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Academic Press: Cambridge, MA, USA, 2011; ISBN 9780123919274. [Google Scholar]
- Ferreira, G.A.; Loh, W. Liquid crystalline nanoparticles formed by oppositely charged surfactant-polyelectrolyte complexes. Curr. Opin. Colloid Interface Sci. 2017, 32, 11–22. [Google Scholar] [CrossRef]
- Guzmán, E.; Fernández-Peña, L.; Ortega, F.; Rubio, R.G. Equilibrium and kinetically trapped aggregates in polyelectrolyte–oppositely charged surfactant mixtures. Curr. Opin. Colloid Interface Sci. 2020, 48, 91–108. [Google Scholar] [CrossRef]
- Exerowa, D.; Kashchiev, D.; Platikanov, D. Stability and permeability of amphiphile bilayers. Adv. Colloid Interface Sci. 1992, 40, 201–256. [Google Scholar] [CrossRef]
- De Gennes, P.-G. Some remarks on coalescence in emulsions or foams. Chem. Eng. Sci. 2001, 56, 5449–5450. [Google Scholar] [CrossRef]
- Ritacco, H.A. Complexity and self-organized criticality in liquid foams. A short review. Adv. Colloid Interface Sci. 2020, 285, 102282. [Google Scholar] [CrossRef] [PubMed]
- Kloek, W.; van Vliet, T.; Meinders, M. Effect of Bulk and Interfacial Rheological Properties on Bubble Dissolution. J. Colloid Interface Sci. 2001, 237, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Meinders, M.B.; van Vliet, T. The role of interfacial rheological properties on Ostwald ripening in emulsions. Adv. Colloid Interface Sci. 2004, 108–109, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Isert, N.; Maret, G.; Aegerter, C.M. Coarsening dynamics of three-dimensional levitated foams: From wet to dry. Eur. Phys. J. E 2013, 36, 116. [Google Scholar] [CrossRef] [Green Version]
- Ritacco, H.A.; Busch, J. Dynamic Surface Tension of Polyelectrolyte/Surfactant Systems with Opposite Charges: Two States for the Surfactant at the Interface. Langmuir 2004, 20, 3648–3656. [Google Scholar] [CrossRef] [PubMed]
- Fameau, A.L.; Arnould, A.; Lehmann, M.; Von Klitzing, R. Photoresponsive self-assemblies based on fatty acids. Chem. Commun. 2015, 51, 2907–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevallier, E.; Monteux, C.; Lequeux, F.; Tribet, C. Photofoams: Remote Control of Foam Destabilization by Exposure to Light Using an Azobenzene Surfactant. Langmuir 2021, 28, 48. [Google Scholar] [CrossRef] [PubMed]
- Honnigfort, C.; Campbell, R.A.; Droste, J.; Gutfreund, P.; Hansen, M.R.; Ravoo, B.J.; Braunschweig, B. Unexpected monolayer-to-bilayer transition of arylazopyrazole surfactants facilitates superior photo-control of fluid interfaces and colloids. Chem. Sci. 2020, 11, 2085–2092. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cs/mM | Total Thickness/Å | ED 1 | Distance from the Air to Maximum ED/Å |
|---|---|---|---|
| 10−3 | 48.9 ± 1 (pH 3.5) 2 | 1.52 (pH 3.5) 2 | 17.1 (pH 3.5) 2 |
| 4 × 10−2 | 52 ± 2 (pH 3.5) | 1.54 (pH 3.5) | 16 (pH 3.5) |
| 0.1 | 44.5 ±1 (pH 3.5) | 1.52 (pH 3.5) | 11.3 (pH 3.5) |
| 25.9 ± 1 (pH 6) | 1.7 (pH 6) | 9.6 (pH 3.5) | |
| 37.6 ±1 (pH 11) | 1.44 (pH 11) | 14.2 (pH 11) | |
| 0.5 | 45.8 ± 1 (pH 3.5) | 1.52 (pH 3.5) | 12.7 (pH 3) |
| 26.8 ± 1 (pH 6) | 1.49 (pH 6) | 13.1 (pH 6) | |
| 28.2 ± 1 (pH 11) | 1.44 (pH 11) | 13.3 (pH 11) |
| Time/Min | Total Thickness/Å | ED 1 | Distance from the Air to Maximum ED/Å |
|---|---|---|---|
| 15 | 15.3 ± 0.8 | 1.18 | 14.1 |
| 55 | 30.0 ± 0.6 | 1.37 | 13.2 |
| 95 | 36.9 ± 0.5 | 1.76 | 12.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, H.; Domínguez, C.; Leyes, M.F.; Moya, S.; Ritacco, H. A pH-Responsive Foam Formulated with PAA/Gemini 12-2-12 Complexes. Colloids Interfaces 2021, 5, 37. https://doi.org/10.3390/colloids5030037
Martinelli H, Domínguez C, Leyes MF, Moya S, Ritacco H. A pH-Responsive Foam Formulated with PAA/Gemini 12-2-12 Complexes. Colloids and Interfaces. 2021; 5(3):37. https://doi.org/10.3390/colloids5030037
Chicago/Turabian StyleMartinelli, Hernán, Claudia Domínguez, Marcos Fernández Leyes, Sergio Moya, and Hernán Ritacco. 2021. "A pH-Responsive Foam Formulated with PAA/Gemini 12-2-12 Complexes" Colloids and Interfaces 5, no. 3: 37. https://doi.org/10.3390/colloids5030037
APA StyleMartinelli, H., Domínguez, C., Leyes, M. F., Moya, S., & Ritacco, H. (2021). A pH-Responsive Foam Formulated with PAA/Gemini 12-2-12 Complexes. Colloids and Interfaces, 5(3), 37. https://doi.org/10.3390/colloids5030037