
Wettability and Stability of Naproxen, Ibuprofen and/or Cyclosporine A/Silica Delivery Systems



Abstract
:1. Introduction
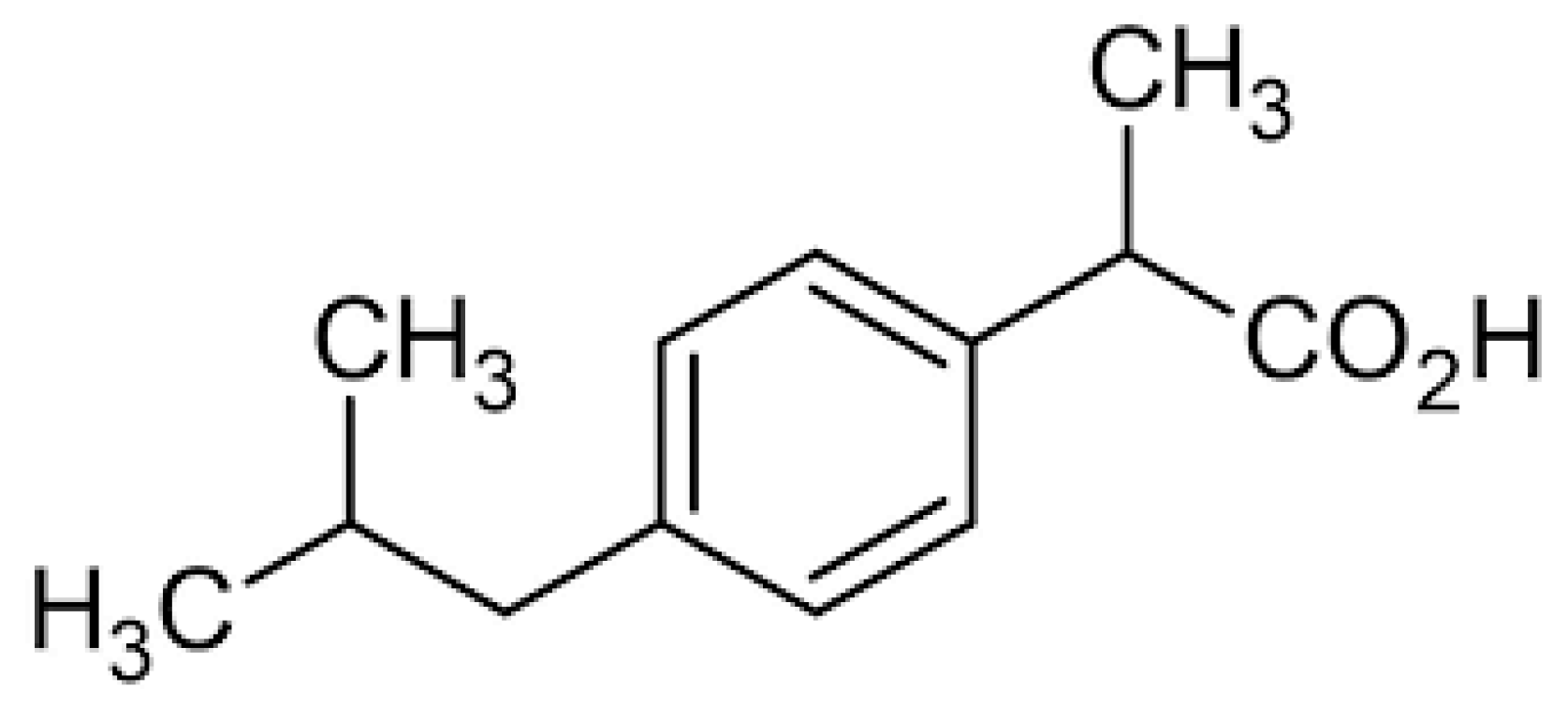
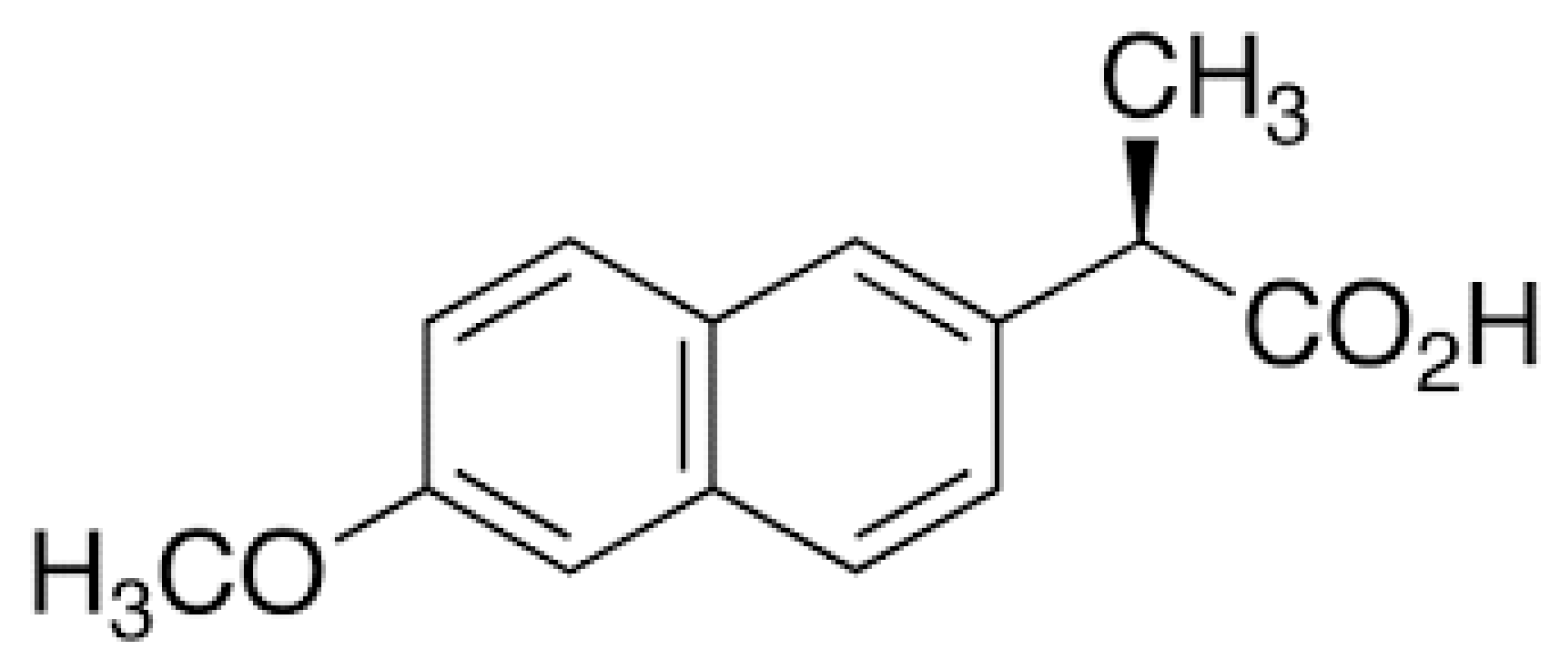
1.1. Non-Steroidal Anti-Inflammatory Drugs
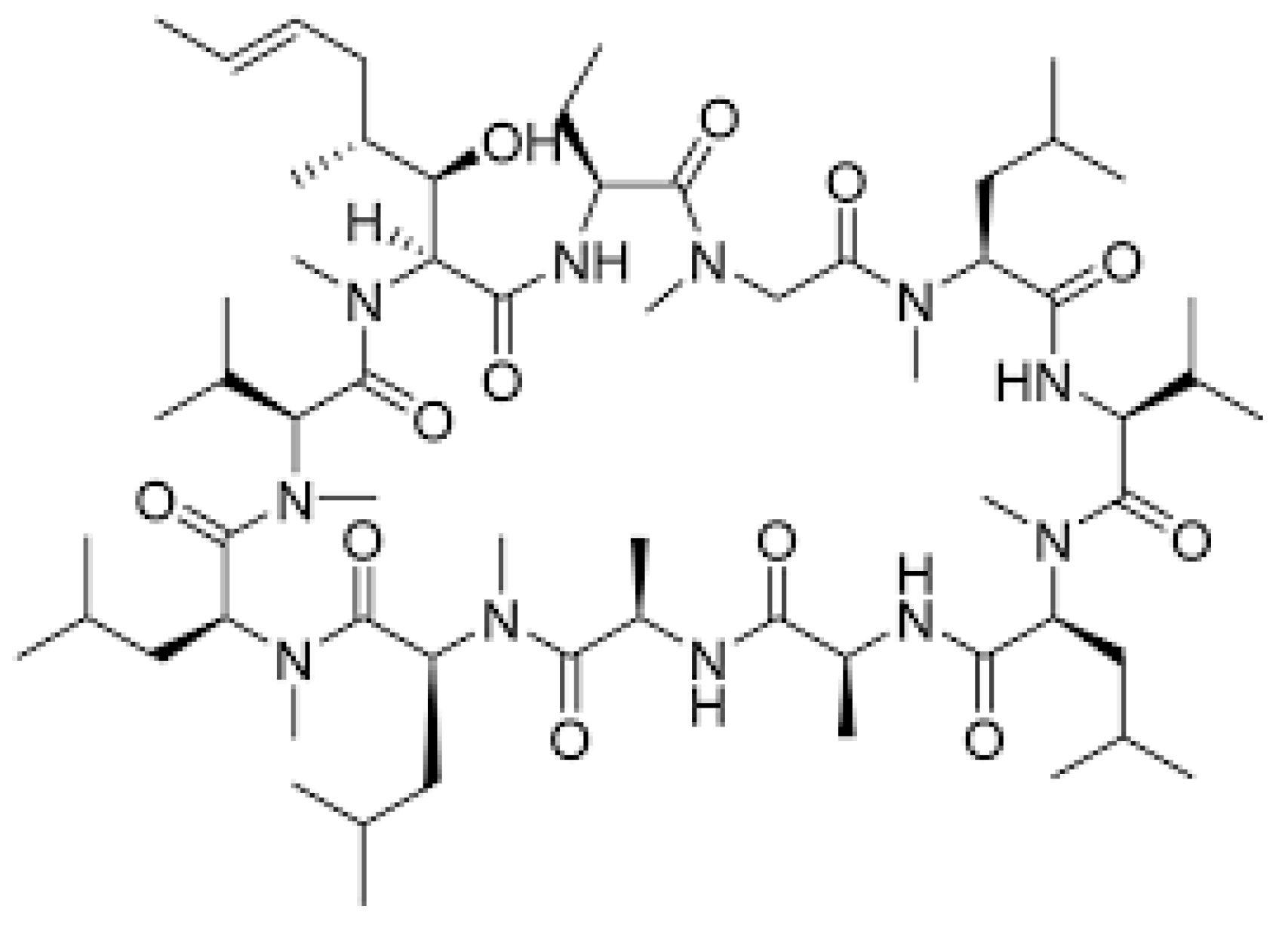
1.2. Immunosuppressive Drugs
1.3. Stability of Drugs
1.4. The Routes of Drug Delivery
1.4.1. Passive Diffusion
1.4.2. Active Transport
1.5. Wettability of Drugs
2. Materials and Methods
2.1. Materials
2.2. Silica-Based Drug Delivery
2.3. Methods
2.3.1. Dynamic Light Scattering
2.3.2. Microelectrophoresis
2.3.3. Thin-Layer Wicking Method
3. Results
3.1. Physico-Chemical Characteristic of Nanocarriers
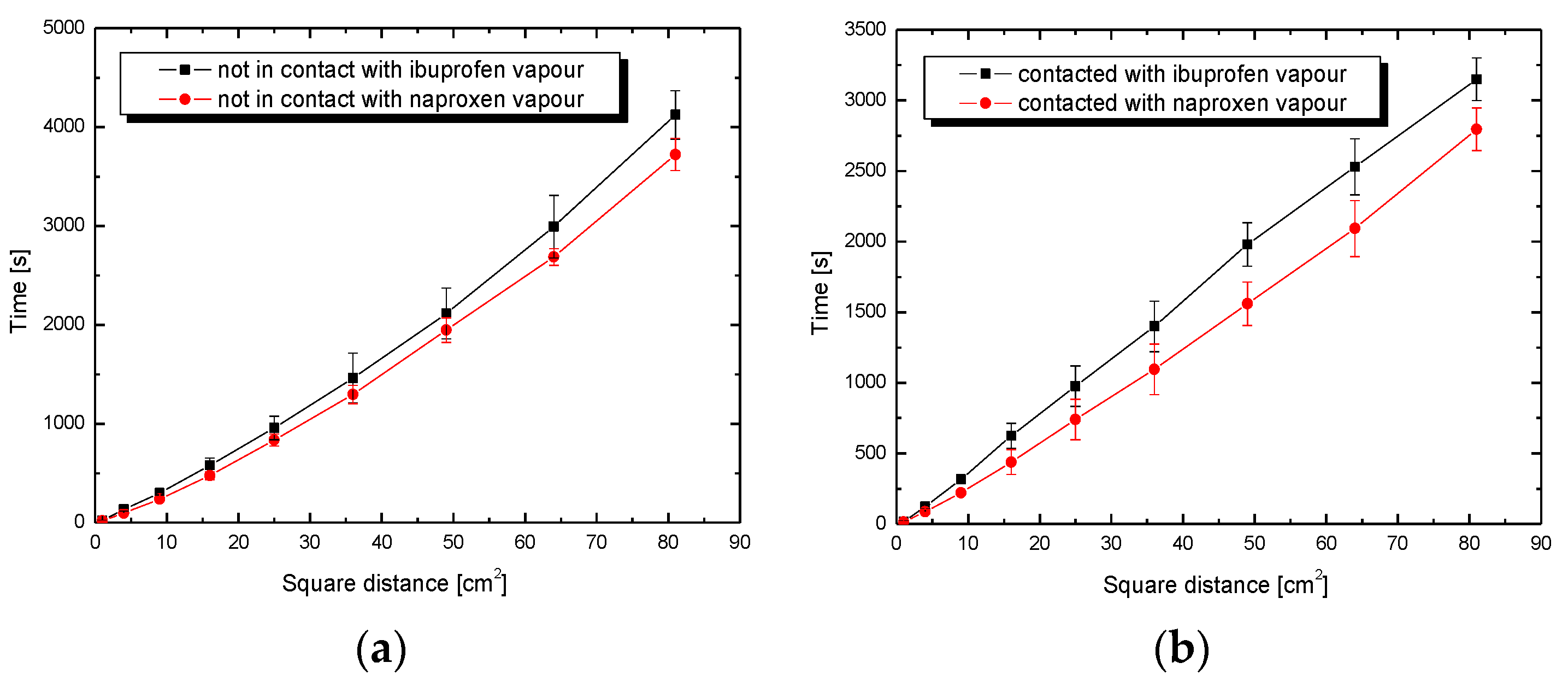
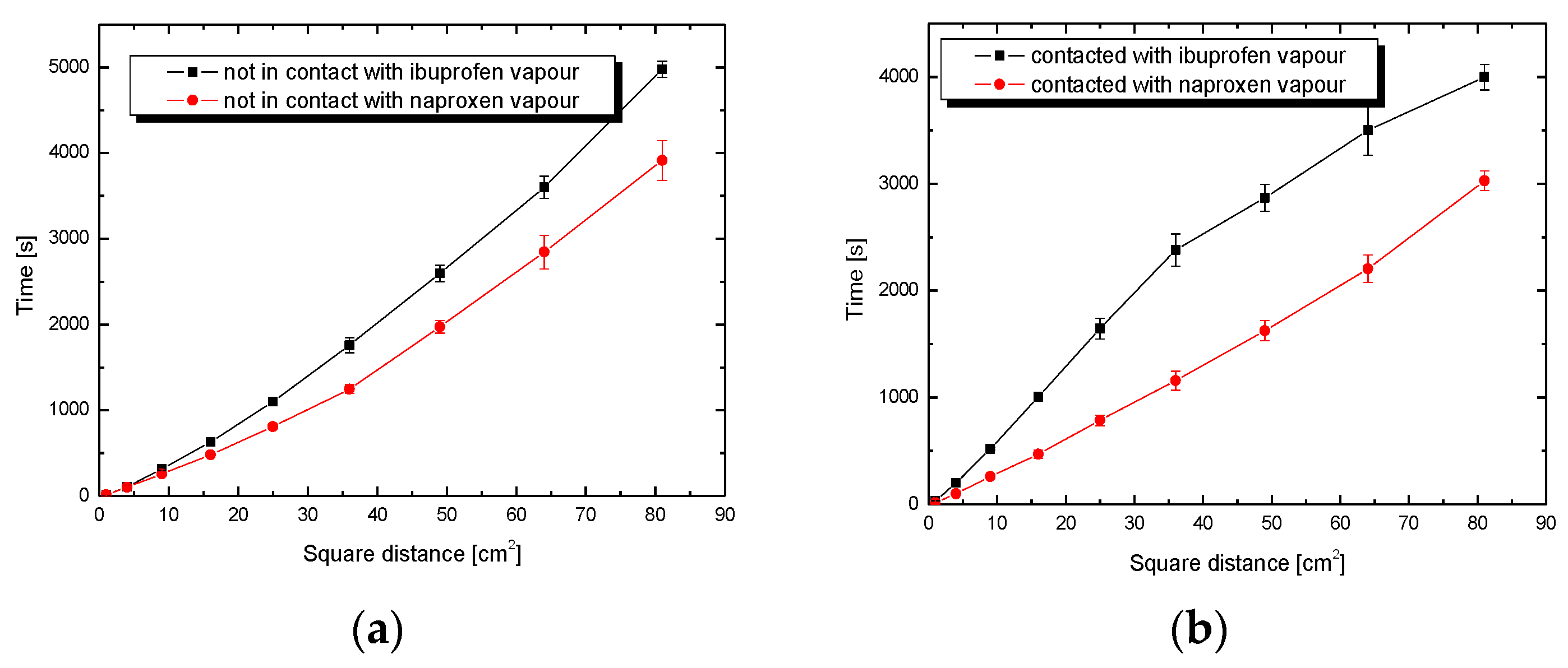
3.2. Wettability Process
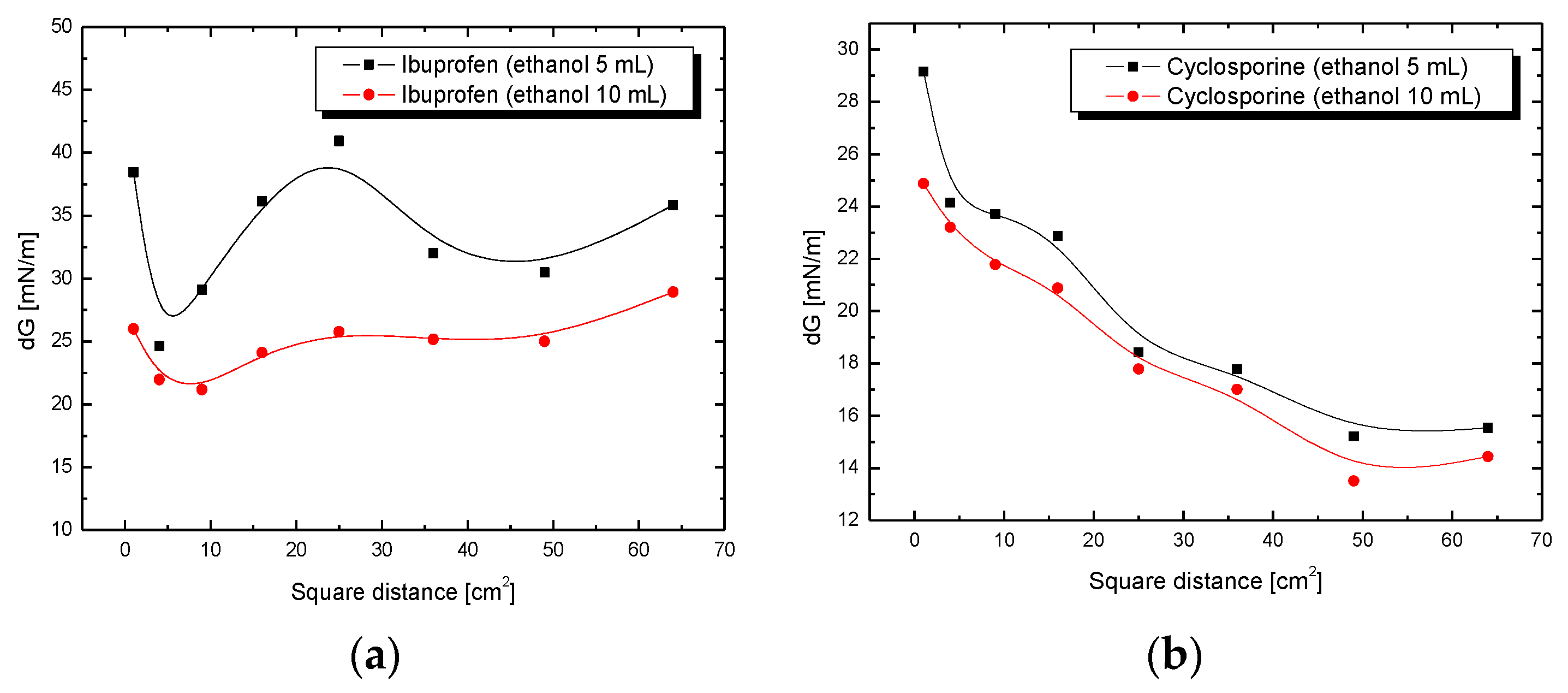
3.3. Adhesion Tension
3.4. The Influence of the Drug Type on the Effective Diameter
3.5. The Influence of the Drug Type on the Zeta Potential
3.6. The Influence of the Drug Type on pH and Polydispersity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Inflammopharmacology 2009, 17, 275–342. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D. Essentials of Pharmaceutical Chemistry, 4th ed.; Pharmaceutical Press: Aberdeen, UK, 2012. [Google Scholar]
- Sun, W.; Hu, Q.; Ji, W.; Wright, G.; Gu, Z. Leveraging Physiology for Precision Drug Delivery. Physiol. Rev. 2017, 97, 189–225. [Google Scholar] [CrossRef]
- Ghlichloo, I.; Gerriets, V. Nonsteroidal Anti-inflammatory Drugs (NSAIDs); Stat Pearls Publishing: Treasure Islands, FL, USA, 2020. [Google Scholar]
- Wiącek, A.E. Changes in wetting properties of silica surface treated with DPPC in the presence of phospholipase A2 enzyme. Appl. Surf. Sci. 2010, 256, 7672–7677. [Google Scholar] [CrossRef]
- Krasucka, P.; Goworek, J. MCM-41 silica as a host material for controlled drug delivery systems. Ann. Univ. Maria Curie-Skłodowska 2015, 70, 45–66. [Google Scholar]
- Wiącek, A.E. Investigation of DPPC adsorption on SiO2 particles from aqueous solution and in the presence of enzymes by the zeta potential and effective diameter measurements. Powder Technol. 2011, 213, 141–147. [Google Scholar] [CrossRef]
- Yoo, J.; Won, Y.-Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, A.; Paiva, A.; Duarte, A.R.C. Therapeutic Liquid Formulations Based on Low Transition Temperature Mixtures for the Incorporation of Anti-Inflammatory Drugs. Pharmaceutics 2021, 13, 1620. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V., Jr.; Ansel, H.C. Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems, 4th ed.; Wolter Kluwer Health: Philadelphia, PA, USA, 2014; pp. 34–66. [Google Scholar]
- Laurano, R.; Boffito, M. Thermosensitive Micellar Hydrogels as Vehicles to Deliver Drugs with Different Wettability. Front. Bioeng. Biotechnol. 2020, 8, 708. [Google Scholar] [CrossRef]
- Colombo, D.; Poggi, S. Clinical profile of cyclosporine in dermatology. Drug Dev. Res. 2011, 72, 634–646. [Google Scholar] [CrossRef]
- Žid, L.; Zeleňák, V.; Almáši, M.; Zeleňáková, A.; Szücsová, J.; Bednarčík, J.; Šuleková, M.; Hudák, A.; Váhovská, L. Mesoporous Silica as a Drug Delivery System for Naproxen: Influence of Surface Functionalization. Molecules 2020, 25, 4722. [Google Scholar] [CrossRef] [PubMed]
- Przykaza, K.; Woźniak, K.; Jurak, M.; Wiącek, A.E.; Mroczka, R. Properties of the Langmuir and Langmuir–Blodgett mono-layers of cholesterol-cyclosporine A on water and polymer support. Adsorption 2019, 25, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Kaaz, K.; Reich, A. Treatment of atopic dermatitis with cyclosporine A: A case report. Dermatol. Rev. 2017, 104, 433. [Google Scholar]
- Wiącek, A.E.; Jurak, M.; Ładniak, A.; Przykaza, K.; Szafran, K. Cyclosporine CsA—The Physicochemical Characterization of Liposomal and Colloidal Systems. Colloids Interfaces 2020, 4, 46. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Singh, R.K.; Perez, R.; Neel, E.A.A.; Kim, H.-W.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Iturrioz-Rodríguez, N.; Correa-Duarte, M.A.; Fanarraga, M.L. Controlled drug delivery systems for cancer based on meso-porous silica nanoparticles. Int. J. Nanomed. 2019, 14, 3389–3401. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Xiao, H.; Li, C.; Dai, Y.; Cheng, Z.; Hou, Z.; Lin, J. Inorganic nanocarriers for platinum drug delivery. Mater. Today 2015, 18, 554–564. [Google Scholar] [CrossRef]
- Salazar-Hernández, C.; Salazar-Hernández, M.; Lona-Ramos, R.; Elorza-Rodríguez, E.; Rocha-Ramírez, A.H.; Carmen, S.-H.; Mercedes, S.-H.; Rocío, L.R.; Enrique, E.-R.; Hilario, R.-R.A. Silica from Rice as New Drug Delivery Systems; IntechOpen Book Series: London, UK, 2017; p. 5. [Google Scholar] [CrossRef] [Green Version]
- Selvarajan, V.; Obuobi, S.; Lai, P.; Ee, R. Silica Nanoparticles—A Versatile Tool for the Treatment of Bacterial Infections. Front. Chem. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Chen, J.-F.; Ding, H.-M.; Wang, J.-X.; Shao, L. Preparation and characterization of porous hollow silica nanoparticles for drug delivery application. Biomaterials 2003, 25, 723–727. [Google Scholar] [CrossRef]
- Liberman, A.; Mendez, N.; Trogler, W.C.; Kummel, A.C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2014, 69, 132–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giret, S.; Chi Man, M.W.; Carcel, C. Mesoporous-Silica-Functionalized Nanoparticles for Drug Delivery. Chemistry 2015, 21, 13850–13865. [Google Scholar] [CrossRef] [PubMed]
- Bharti, C.; Nagaich, U.; Pal, A.K.; Gulati, N. Mesoporous silica nanoparticles in target drug delivery system: A review. Int. J. Pharm. Investig. 2015, 5, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiącek, A.E. Investigations of DPPC effect on Al2O3 particles in the presence of (phospho)lipases by the zeta potential and effective diameter measurements. Appl. Surf. Sci. 2011, 257, 4495–4504. [Google Scholar] [CrossRef]
- Kralchevsky, P.A.; Danov, K.D.; Denkov, N.D. Chemical physics of colloid systems and interfaces. In Handbook of Surface and Colloid Chemistry, 3rd ed.; Birdi, K.S., Ed.; CRC Press: Boca Raton, FL, USA, 2008; Volume 7, pp. 197–377. [Google Scholar]
- Otsuki, A.; de Campo, L.; Garvey, C.; Rehm, C. H2O/D2O Contrast Variation for Ultra-Small-Angle Neutron Scattering to Minimize Multiple Scattering Effects of Colloidal Particle Suspensions. Colloids Interfaces 2018, 2, 37. [Google Scholar] [CrossRef] [Green Version]
- Guide for Dynamic Light Scattering (DLS) Sample Preparation. Available online: https://www.brookhaveninstruments.com/guide-for-dls-sample-preparation/08.11.2019 (accessed on 27 December 2021).
- Otsuki, A.; Hayagan, N.L. Zeta potential of inorganic fine particle-Na-bentonite binder mixture systems. Electrophoresis 2020, 41, 1405–1412. [Google Scholar] [CrossRef]
- Vergouw, J.; Difeo, A.; Xu, Z.; Finch, J. An agglomeration study of sulphide minerals using zeta-potential and settling rate. Part 1: Pyrite and galena. Miner. Eng. 1998, 11, 159–169. [Google Scholar] [CrossRef]
- Suchata, K.; Muenduen, P.; Zhang, N.B.-M. Applicability of Washburn capillary rise for determining contact angles of powders/porous materials. J. Colloid Interface Sci. 2013, 397, 169–176. [Google Scholar]
- Wiącek, A.E. Changes in wetting properties of alumina surface treated with DPPC in the presence of phospholipase A2 enzyme. Colloids Surf. B Biointerfaces 2011, 87, 54–60. [Google Scholar] [CrossRef]
- Chibowski, E. Some problems of characterization of a solid surface via the surface free energy changes. Adsorption Sci. Technol. 2017, 35, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Terpiłowski, K.; Bielska, M.; Prochaska, K.; Chibowski, E. Wettability of ultrafiltration membranes. In Advances in Contact Angle, Wettability and Adhesion; Mittal, K.L., Ed.; Wiley Online Library: Hoboken, NJ, USA, 2019; Volume 4, pp. 57–72. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Routes of Drug Delivery to the Body | Forms of the Drug |
|---|---|
| oral and sublingual route | tablets (effervescent, coated, dragées), microcapsules, capsules, granules, powders for oral suspension preparation, syrups, two-phase tablets |
| injection (subcutaneous, intramuscular, intravascular, rectal) | suppositories, injections |
| respiratory tract | aerosols, inhalations |
| percutaneous | ointments, foams, gels, creams, transdermal patches |
| Time (min.) | Effective Diameter of Naproxen Solution (nm) | Effective Diameter of Ibuprofen Solution (nm) | Effective Diameter of Cyclosporine Solution (nm) |
|---|---|---|---|
| 5 | 320.1 | 232.5 | 30035.1 |
| 15 | 411.5 | 325.9 | 28358.0 |
| 30 | 529.0 | 451.0 | 26681.1 |
| 60 | 572.2 | 1157.6 | 25393.9 |
| 120 | 1333.9 | 1238.7 | 16501.5 |
| Time (min.) | Zeta Potential of Naproxen Solution (mV) | Zeta Potential of Ibuprofen Solution (mV) | Zeta Potential of Cyclosporine Solution (mV) |
|---|---|---|---|
| 5 | 1.840 ± 0.8 | 0.964 ± 0.7 | 5.53 ± 3.6 |
| 15 | 2.220 ± 1.9 | −0.195 ± 0.3 | 0.15 ± 0.5 |
| 30 | 0.223 ± 0.4 | 2.140 ± 3.5 | 0.49 ± 0.2 |
| 60 | 0.470 ± 3.3 | −4.070 ± 5.0 | 1.96 ± 2.3 |
| 120 | 0.200 ± 0.5 | −1.360 ± 1.1 | 2.46 ± 2.4 |
| Time (min.) | Polydispersity of Naproxen Solution | Polydispersity of Ibuprofen Solution | Polydispersity of Cyclosporine Solution |
|---|---|---|---|
| 5 | 0.270 | 0.488 | 0.917 |
| 15 | 0.340 | 0.577 | 1.010 |
| 30 | 0.313 | 0.460 | 1.258 |
| 60 | 0.338 | 0.598 | 1.146 |
| 120 | 0.305 | 0.283 | 3.225 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiącek, A.E.; Przykaza, K. Wettability and Stability of Naproxen, Ibuprofen and/or Cyclosporine A/Silica Delivery Systems. Colloids Interfaces 2022, 6, 11. https://doi.org/10.3390/colloids6010011
Wiącek AE, Przykaza K. Wettability and Stability of Naproxen, Ibuprofen and/or Cyclosporine A/Silica Delivery Systems. Colloids and Interfaces. 2022; 6(1):11. https://doi.org/10.3390/colloids6010011
Chicago/Turabian StyleWiącek, Agnieszka Ewa, and Kacper Przykaza. 2022. "Wettability and Stability of Naproxen, Ibuprofen and/or Cyclosporine A/Silica Delivery Systems" Colloids and Interfaces 6, no. 1: 11. https://doi.org/10.3390/colloids6010011
APA StyleWiącek, A. E., & Przykaza, K. (2022). Wettability and Stability of Naproxen, Ibuprofen and/or Cyclosporine A/Silica Delivery Systems. Colloids and Interfaces, 6(1), 11. https://doi.org/10.3390/colloids6010011