Ccr6 Deficiency Attenuates Spontaneous Chronic Colitis in Winnie

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Phenotypic Assessment
2.3. Morphological Assessment of Other Organs
2.4. Flowcytometry
2.5. Histology
2.6. Alcian Blue Staining of Mucus Producing Goblet Cells
2.7. Immunohistochemistry
2.8. Real Time Polymerase Chain Reaction (RT-PCR)
2.9. Statistical Data Analysis
3. Results
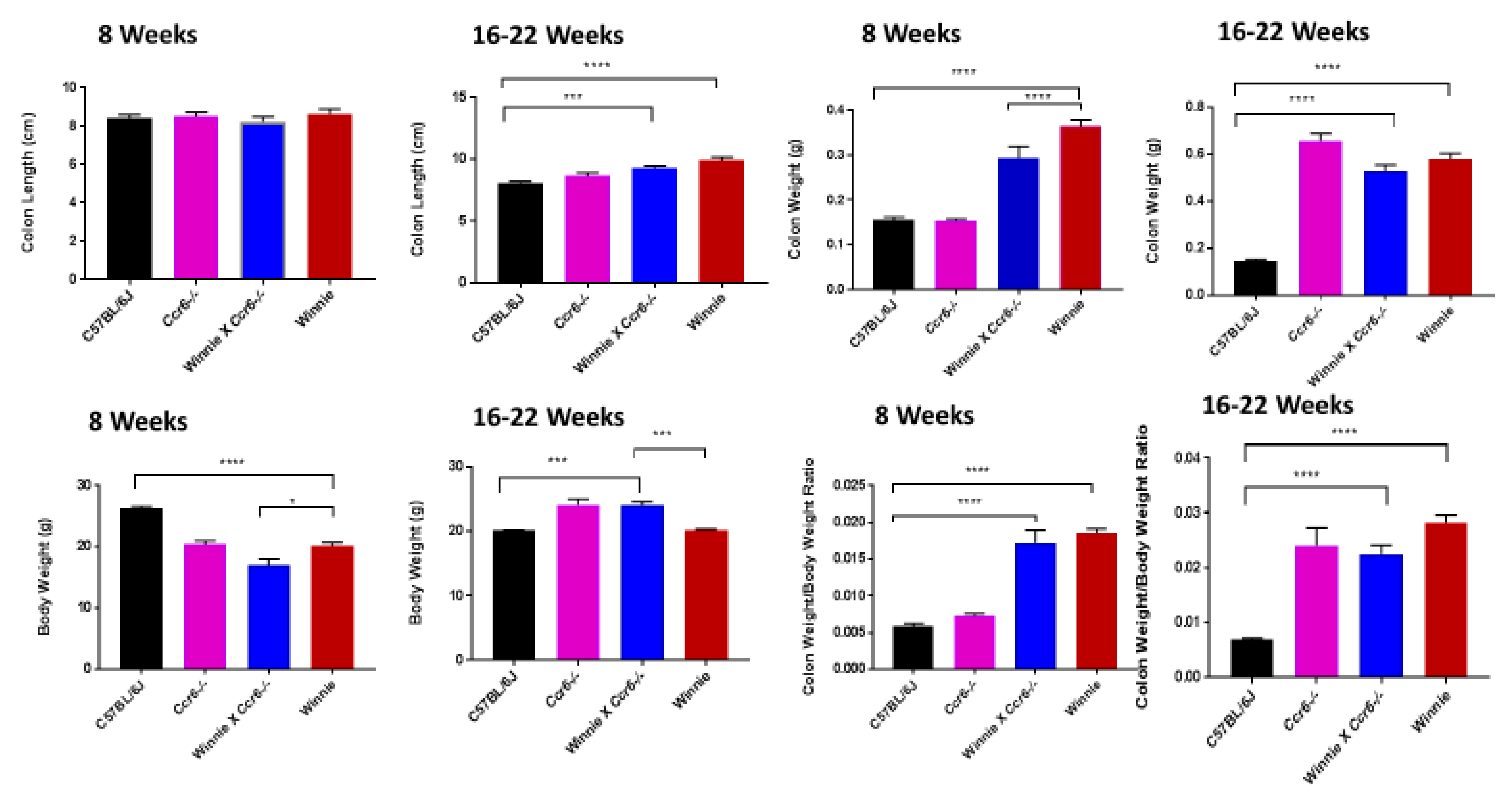
3.1. Clinical Parameters Indicate that Ccr6-Deficiency Lessens Inflammation in the Colon
3.1.1. Clinical Parameters
3.1.2. Disease Activity Index
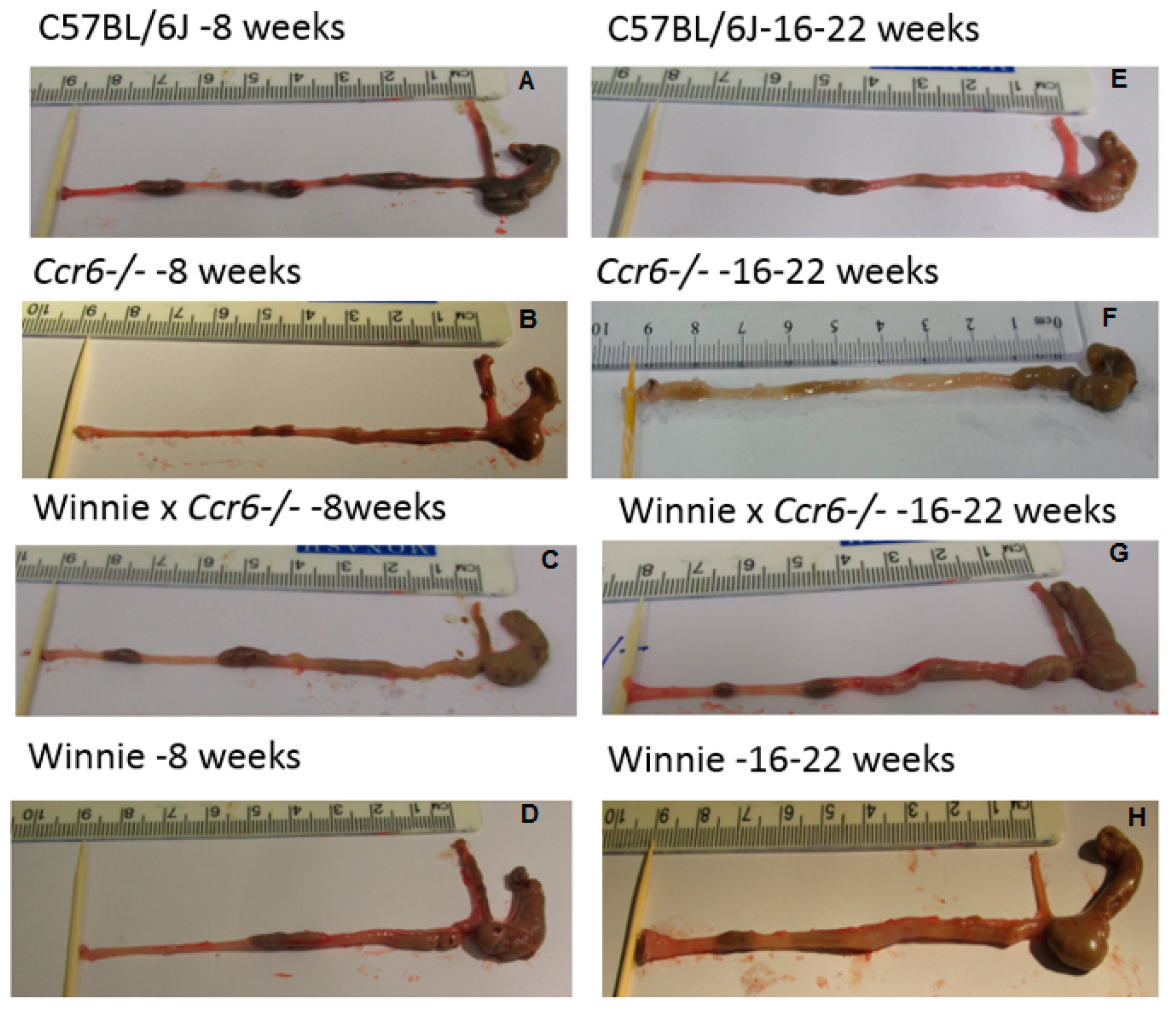
3.1.3. Gross Colon Morphology
3.2. Ccr6-Deficiency Displayed Attenuated Inflammationin Multi-System Pathology Concomitant with Colitis
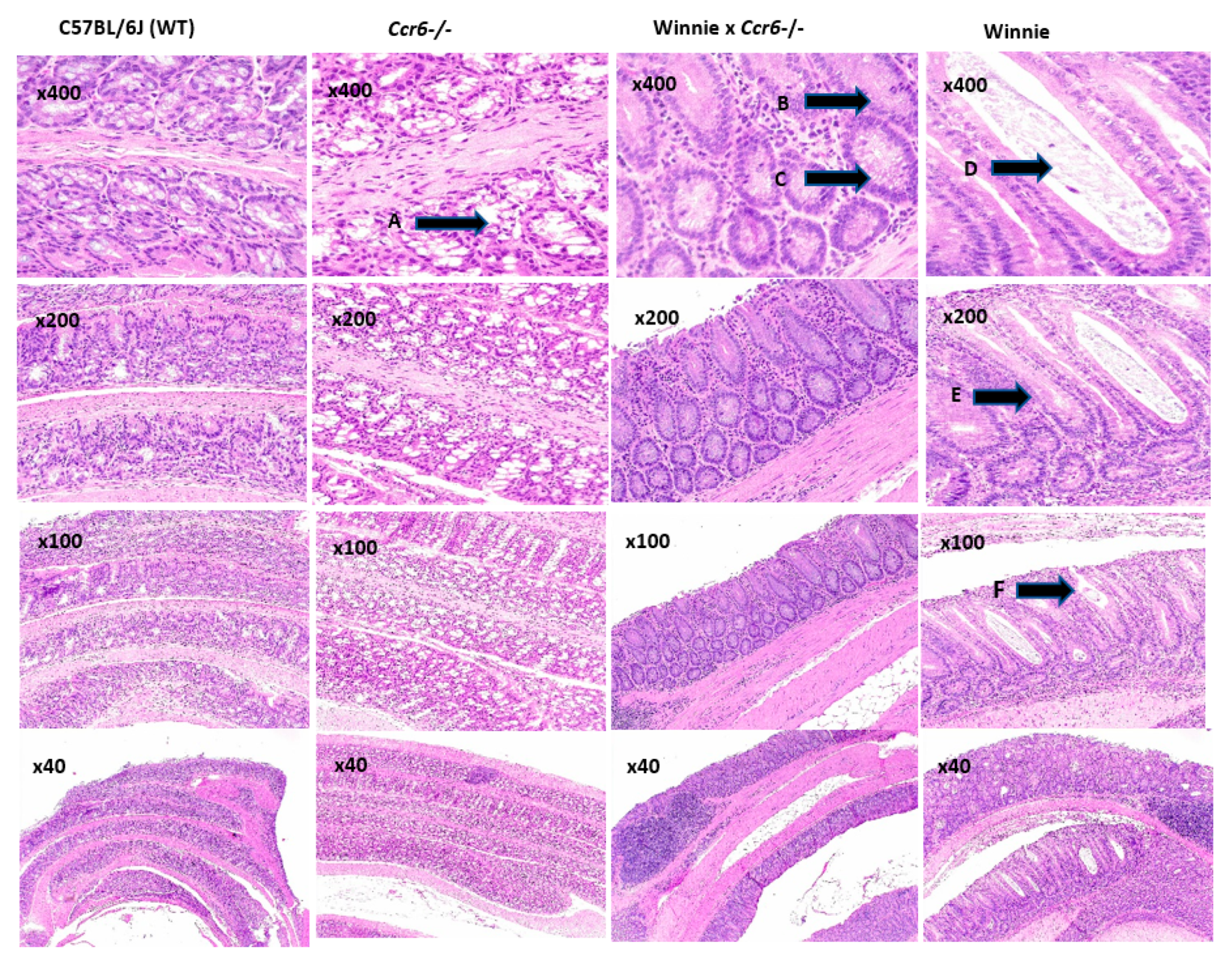
3.2.1. Colon Histology
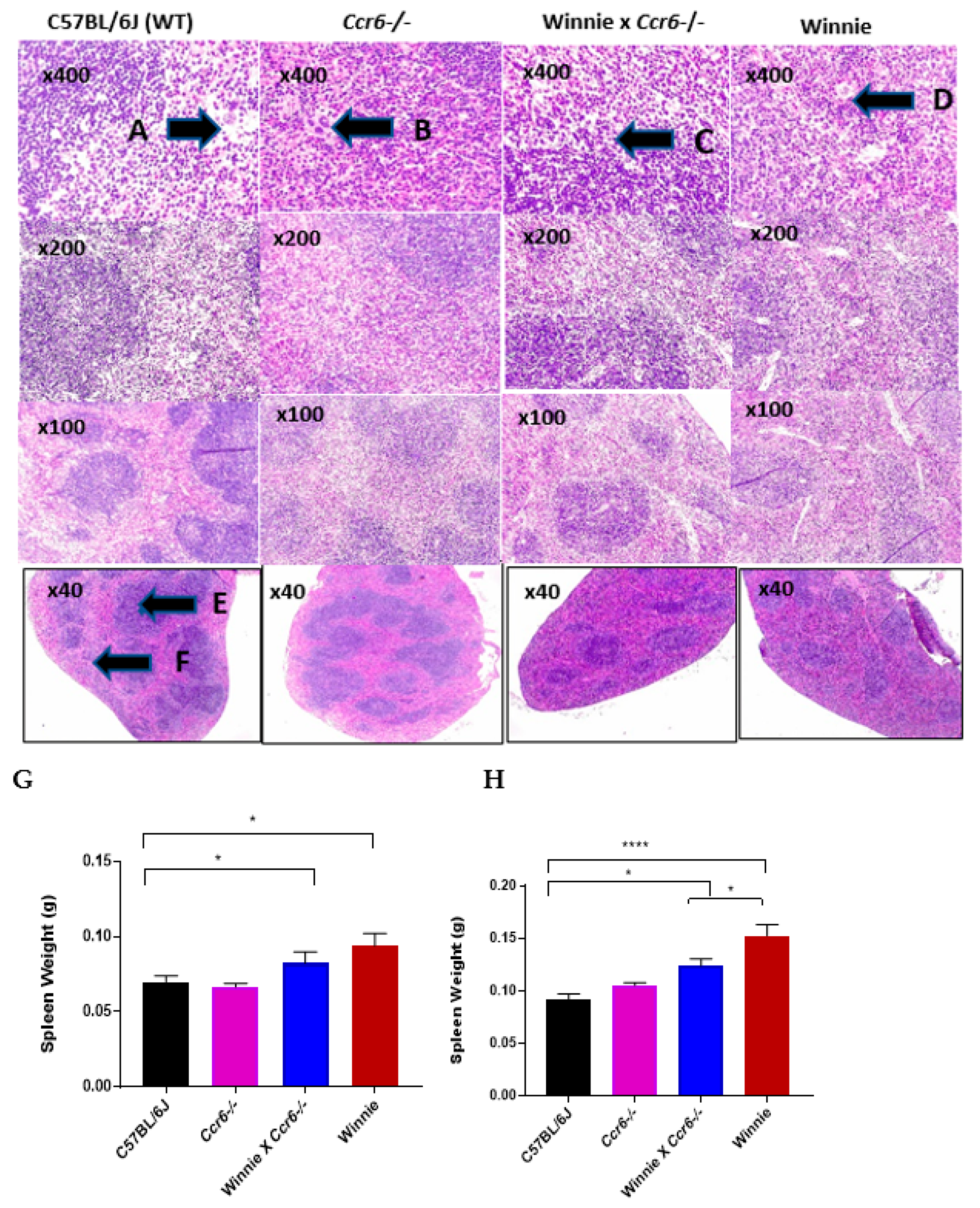
3.2.2. Spleen Weight
3.2.3. Spleen Histology
3.2.4. Liver Weight
3.2.5. Liver Histology
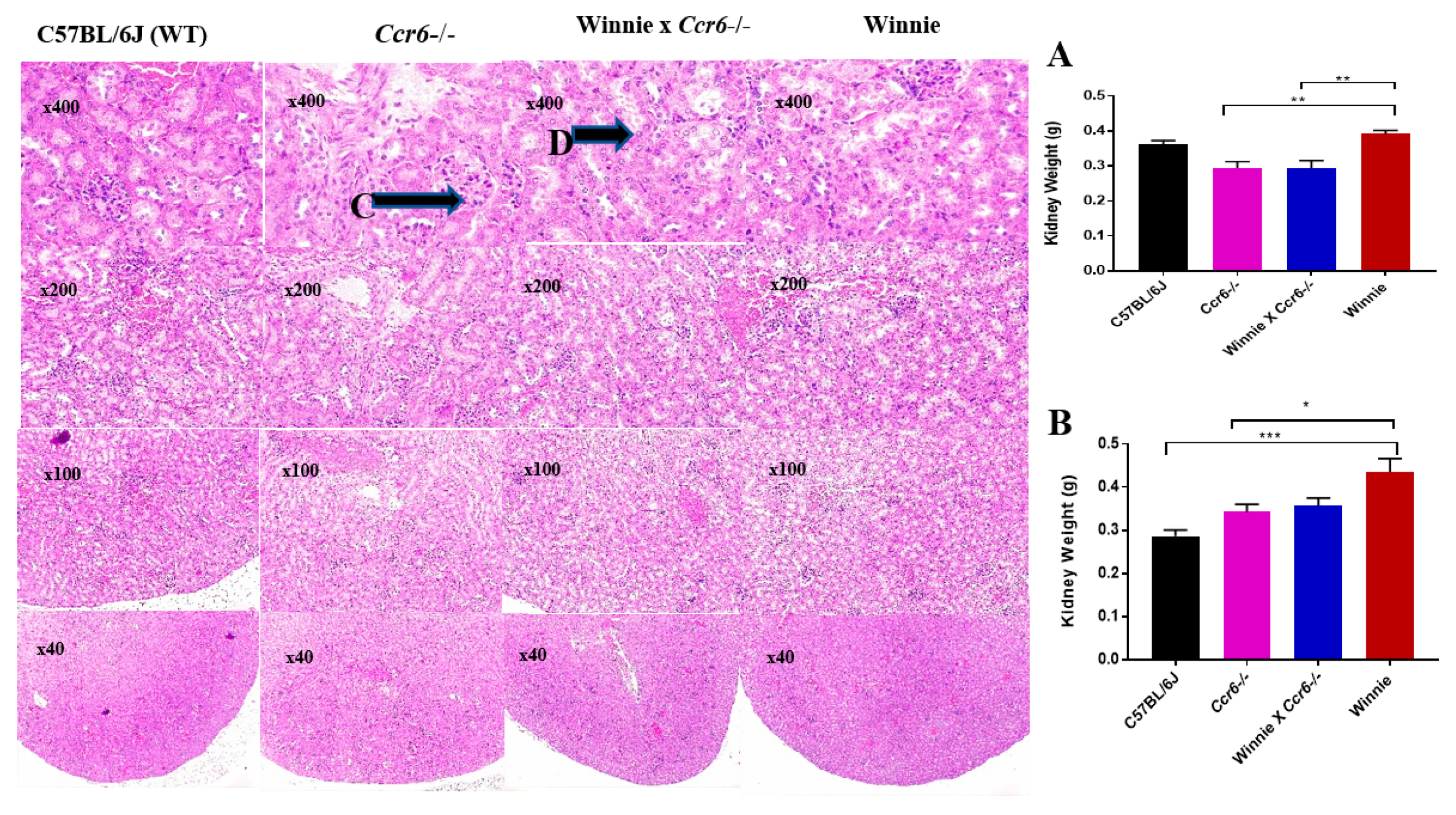
3.2.6. Kidney Weight
3.2.7. Kidney Histology
3.3. Ccr6-Deficiency Reduces the Lymphocyte Distribution in the Spleen and MLN during Colitis
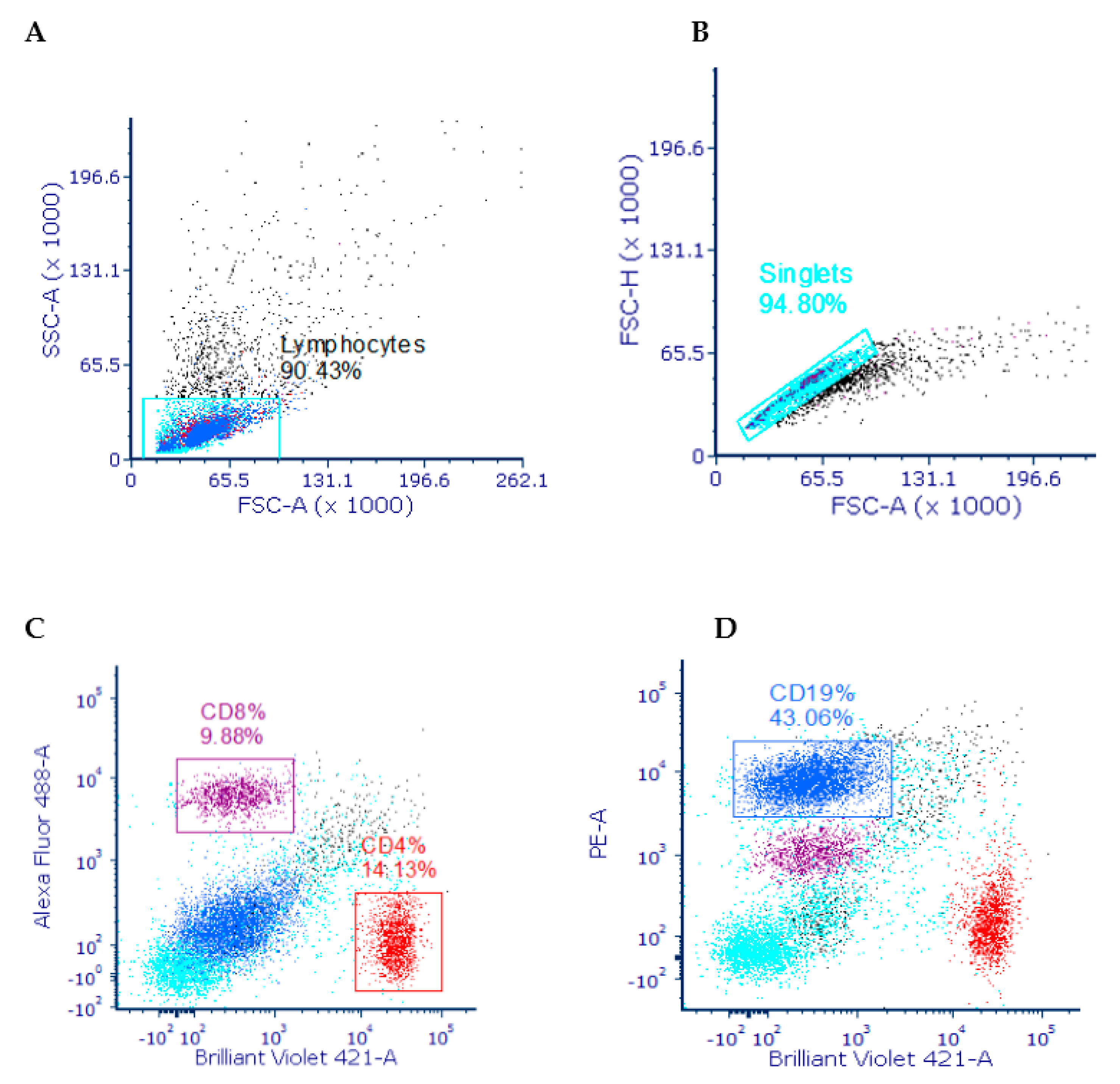
3.3.1. Gating Strategy
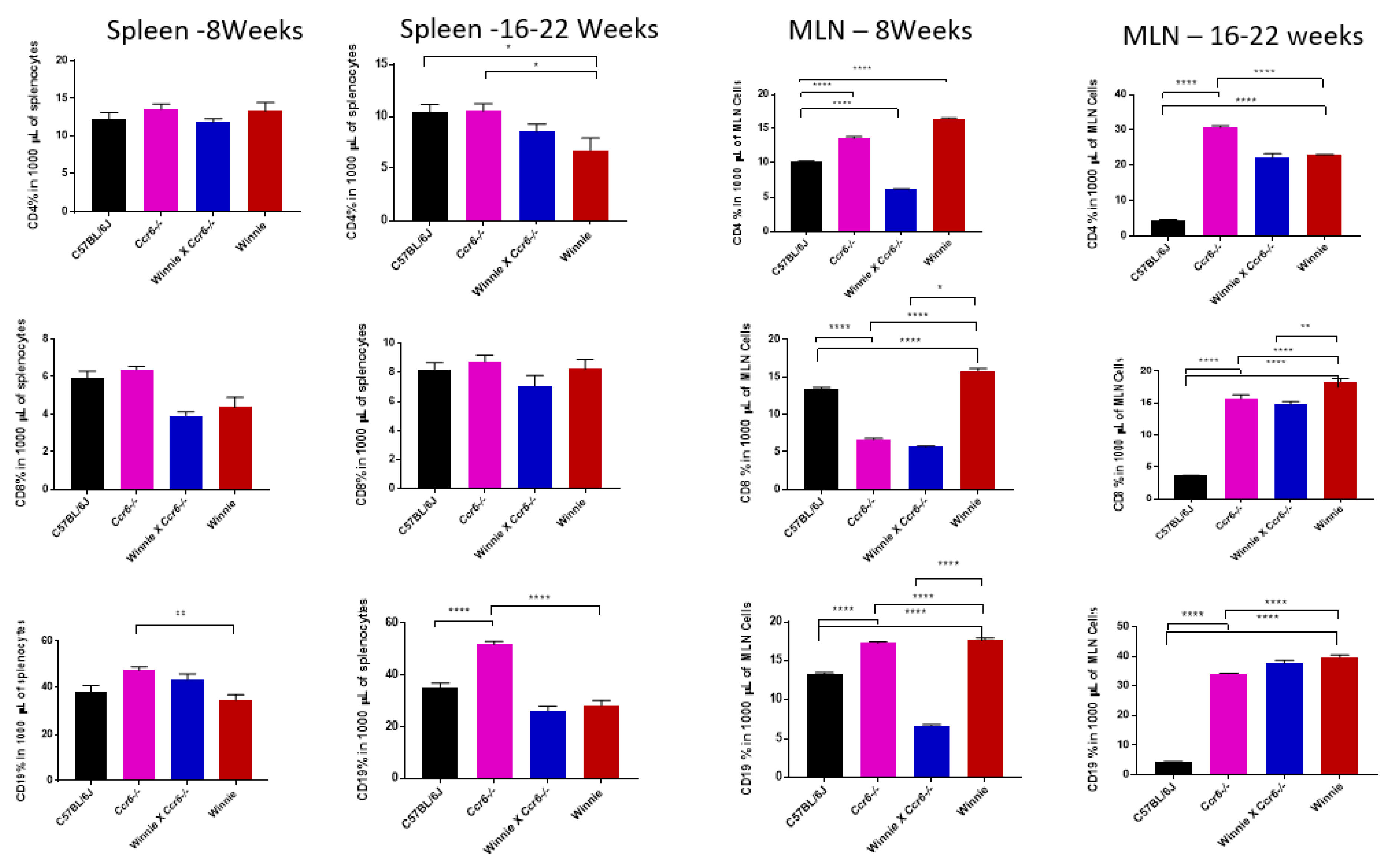
3.3.2. T (CD4 & CD8) and B (CD19) Lymphocyte Distribution in the Spleen
3.3.3. T (CD4 & CD8) and B (CD19) Lymphocyte Distribution in the Mesenteric Lymph Nodes (MLN)
3.4. Ccr6-Deficiency Downregulates Molecular Signalling by CCL20 and PI3KP, Upregulates AktP
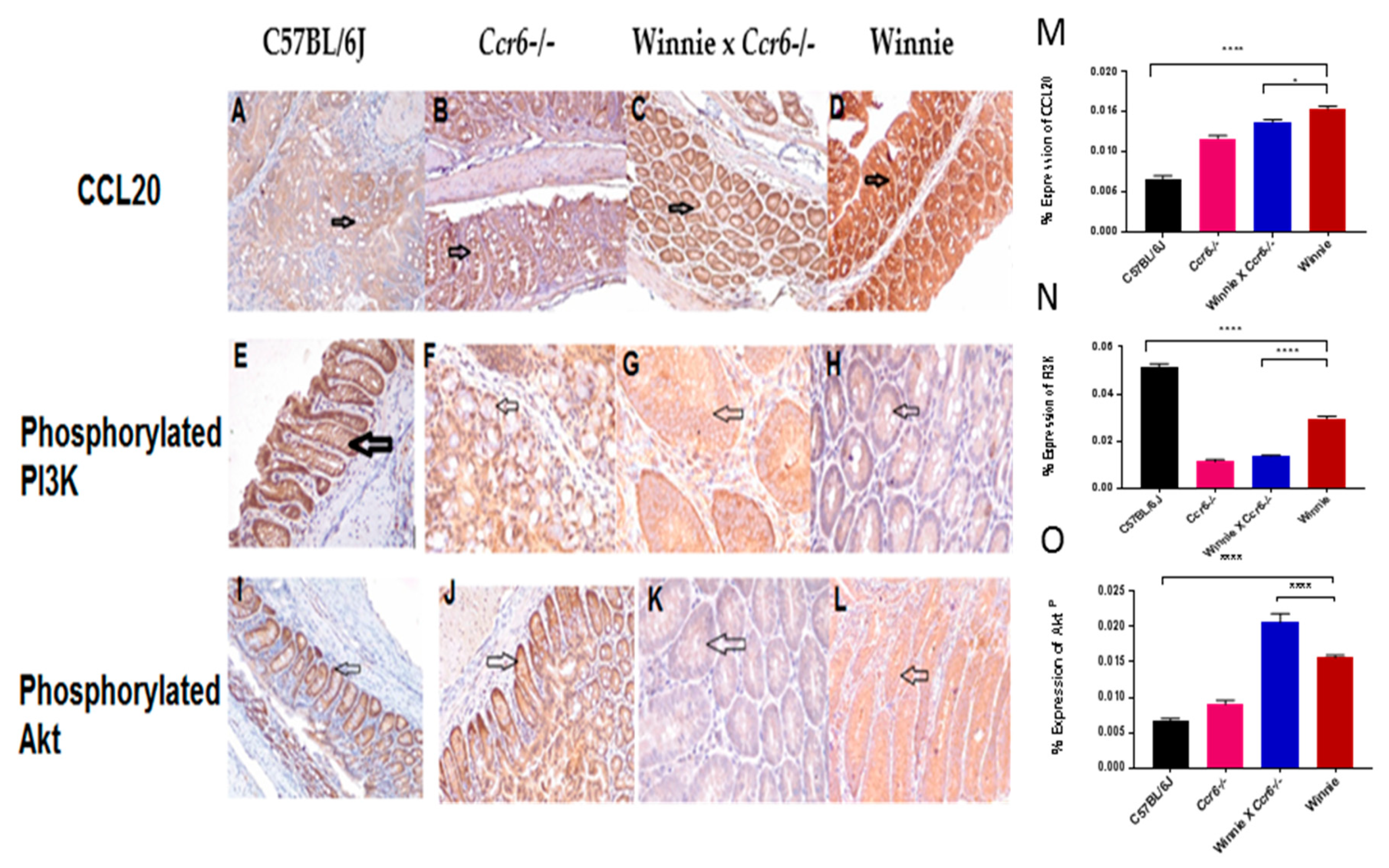
3.4.1. CCL20 Expression Pattern in the Colon
3.4.2. Phosphorylated Phosphoinositide 3-Kinase (PI3KP) Expression Pattern in the Colon
3.4.3. Phosphorylated Akt (AktP) Expression Pattern in the Colon
3.5. Ccr6-Deficiency Suppress the T Helper Lymphocyte Immune Responses During Colitis and Upregulates IL-10
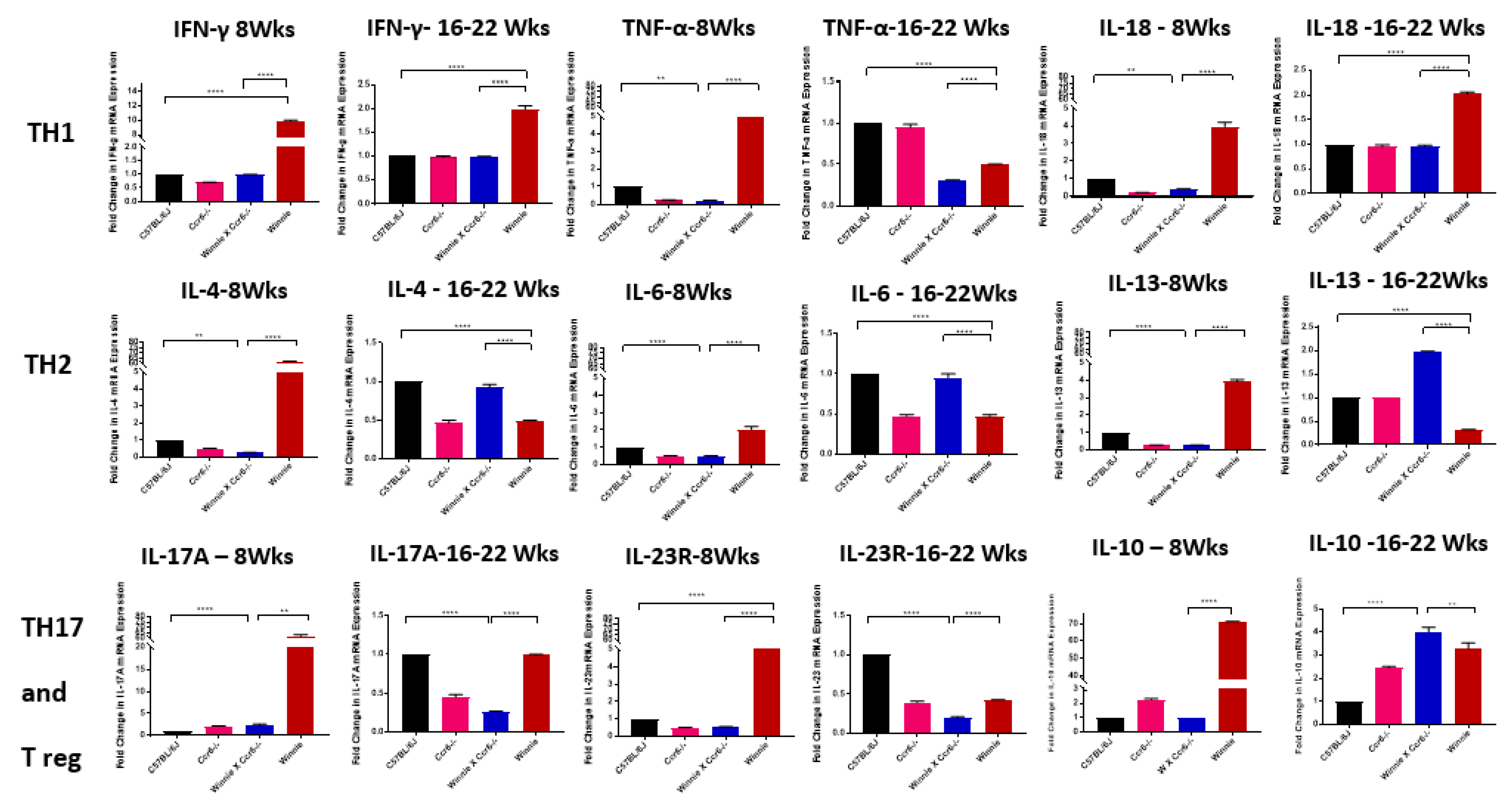
3.5.1. TH1 Cytokine mRNA Expression in the Colon
3.5.2. TH2 Cytokine mRNA Expression in the Colon
3.5.3. TH17 Cytokine mRNA Expression in the Colon
3.5.4. Anti-Inflammatory (Treg) Cytokine mRNA Expression in the Colon
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ranasinghe, R.; Eri, R. CCR6–CCL20 Axis in IBD: What Have We Learnt in the Last 20 Years? Gastrointest. Disord. 2019, 1, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.; Chan, F.K. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Plevinsky, J.; Gumidyala, A.; Fishman, L. Transition experience of young adults with inflammatory bowel diseases (IBD): a mixed methods study. Child 2015, 41, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Ehrlich, O.G.; Allen, J.I.; Meadows, P.; Szigethy, E.M.; Henrichsen, K.; Kim, S.C.; Lawton, R.C.; Murphy, S.M.; Regueiro, M. The Cost of Inflammatory Bowel Disease: An Initiative From the Crohn’s & Colitis Foundation. Inflammat. Bowel Dis. 2019, 26, 1–10. [Google Scholar]
- Sewitch, M.J.; Abrahamowicz, M.; Bitton, A.; Daly, D.; Wild, G.E.; Cohen, A.; Katz, S.; Szego, P.L.; Dobkin, P.L. Psychological distress, social support, and disease activity in patients with inflammatory bowel disease. Am. J. Gastroent. 2001, 96, 1470. [Google Scholar] [CrossRef]
- Hrabe, J.E.; Byrn, J.C.; Button, A.M.; Zamba, G.K.; Kapadia, M.R.; Mezhir, J.J. A matched case-control study of IBD-associated colorectal cancer: IBD portends worse outcome. J. Surg. Oncol. 2014, 109, 117–121. [Google Scholar] [CrossRef]
- Basheer, W.; Kunde, D.; Eri, R. Role of Chemokine Ligand CCL20 and its Receptor CCR6 in lntestinal Inflammation. Immunol. Infect. Dis. 2013, 1, 30–37. [Google Scholar]
- Ranasinghe, R.; Eri, R. CCR6–CCL20-Mediated Immunologic Pathways in Inflammatory Bowel Disease. Gastrointest. Disord. 2019, 1, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Basheer, W. Genetic Ablation of CCR6 Confers Differential Exacerbation in a Spontaneous Colitis Model. Ph.D. Thesis, University of Tasmania, Hobart, Australia, 2018. [Google Scholar]
- Lee, A.; Eri, R.; Lyons, A.B.; Grimm, M.; Korner, H. CC chemokine ligand 20 and its cognate receptor CCR6 in mucosal T cell immunology and inflammatory bowel disease: Odd couple or axis of evil? Front. Immunol. 2013, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, R.; Eri, R. Modulation of the CCR6-CCL20 Axis: A Potential Therapeutic Target in Inflammation and Cancer. Medicina 2018, 54, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateer, S.W.; Cardona, J.; Marks, E.; Goggin, B.J.; Hua, S.; Keely, S. Ex vivo intestinal sacs to assess mucosal permeability in models of gastrointestinal disease. J. Visual. Exper. 2016, e53250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.-E.; Paust, H.-J.; Steinmetz, O.M.; Peters, A.; Riedel, J.-H.; Erhardt, A.; Wegscheid, C.; Velden, J.; Fehr, S.; Mittrücker, H.-W. CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh-Bacic, D.; Lindenmeyer, M.; Cohen, C.D.; Draganovici, D.; Mandelbaum, J.; Edenhofer, I.; Ziegler, U.; Regele, H.; Wüthrich, R.P.; Segerer, S. Expression of the chemokine receptor CCR6 in human renal inflammation. Nephr. Dialysis Transplant. 2010, 26, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oo, Y.H.; Banz, V.; Kavanagh, D.; Liaskou, E.; Withers, D.R.; Humphreys, E.; Reynolds, G.M.; Lee-Turner, L.; Kalia, N.; Hubscher, S.G. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J. Hepat. 2012, 57, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- Arsent’eva, N.; Semenov, A.; Lyubimova, N.; Ostankov, Y.V.; Elezo, D.; Kudryavtsev, I.; Basina, V.; Esaulenko, E.; Kozlov, K.; Zhdanov, K. Chemokine receptors CXCR3 and CCR6 and their ligands in the liver and blood of patients with chronic hepatitis C. Bull. Exper. Biol. Med. 2015, 160, 252–255. [Google Scholar] [CrossRef]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef] [Green Version]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.M.; Rahman, A.A.; Carbone, S.E.; Randall-Demllo, S.; Filippone, R.; Bornstein, J.C.; Eri, R.; Nurgali, K. Alterations of colonic function in the Winnie mouse model of spontaneous chronic colitis. Am. J. Phys. Gastrointest. Liver Physiol. 2016, 312, G85–G102. [Google Scholar] [CrossRef] [Green Version]
- Ranasinghe, R.; Eri, R. Pleiotropic immune functions of chemokine receptor 6 in health and disease. Medicines 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, N.; Meitei, H.T.; Sonar, S.A.; Sharma, P.K.; Mujeeb, V.R.; Srivastava, S.; Boppana, R.; Lal, G. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J. Autoimm. 2018, 88, 121–130. [Google Scholar] [CrossRef]
- Cesta, M.F. Normal structure, function, and histology of the spleen. Toxic. Path. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Doucet, M.; Jayaraman, S.; Swenson, E.; Tusing, B.; Weber, K.L.; Kominsky, S.L. CCL20/CCR6 signaling regulates bone mass accrual in mice. J. Bone Mineral Res. 2016, 31, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Carson IV, W.F.; Cavassani, K.A.; Connett, J.M.; Kunkel, S.L. CCR6 as a mediator of immunity in the lung and gut. Exper. Cell Res. 2011, 317, 613–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, S.; Kohen, R.; Bass, P.; Rubinstein, A. Relation between colonic inflammation severity and total low-molecular-weight antioxidant profiles in experimental colitis. Digest. Dis. Sci. 2000, 45, 1180–1187. [Google Scholar] [CrossRef]
- Katchar, K.; Kelly, C.P.; Keates, S.; O’Brien, M.J.; Keates, A.C. MIP-3α neutralizing monoclonal antibody protects against TNBS-induced colonic injury and inflammation in mice. Am. J. Phys. Gastroint. Liver Physiol. 2007, 292, G1263–G1271. [Google Scholar] [CrossRef]
- Paust, H.-J.; Turner, J.-E.; Riedel, J.-H.; Disteldorf, E.; Peters, A.; Schmidt, T.; Krebs, C.; Velden, J.; Mittrücker, H.-W.; Steinmetz, O.M. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2012, 82, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Egresi, A.; Kovács, Á.; Szilvás, Á.; Blázovics, A. Gut-liver axis in inflammatory bowel disease. A retrospective study. Orv. Hetil. 2017, 158, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.; Brito, B.S.; Silva, I.N.d.N.; Nóbrega, V.G.; da Silva, M.C.S.; Gomes, H.D.d.N.; Fortes, F.M.; Pimentel, A.M.; Mota, J.; Almeida, N. Frequency of Hepatobiliary Manifestations and Concomitant Liver Disease in Inflammatory Bowel Disease Patients. BioMed Res. Int. 2019, 7604939. [Google Scholar] [CrossRef] [Green Version]
- Harmatz, A. Hepatobiliary manifestations of inflammatory bowel disease. Med. Clin. N. Am. 1994, 78, 1387–1398. [Google Scholar] [CrossRef]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Ann. Rev. Immun. 2012, 30, 69–94. [Google Scholar] [CrossRef]
- Ito, R.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Kita, M.; Ueda, Y.; Iwakura, Y. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin. Exper. Immun. 2006, 146, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, F.; Kojouharoff, G.; Hans, W.; Schölmerich, J.; Gross, V.; Falk, W. Interferon-gamma (IFN-γ)-and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exper. Immun. 1999, 116, 238. [Google Scholar] [CrossRef] [PubMed]
- Nava, P.; Koch, S.; Laukoetter, M.G.; Lee, W.Y.; Kolegraff, K.; Capaldo, C.T.; Beeman, N.; Addis, C.; Gerner-Smidt, K.; Neumaier, I. Interferon-γ regulates intestinal epithelial homeostasis through converging β-catenin signaling pathways. Immunity 2010, 32, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thelemann, C.; Eren, R.O.; Coutaz, M.; Brasseit, J.; Bouzourene, H.; Rosa, M.; Duval, A.; Lavanchy, C.; Mack, V.; Mueller, C. Interferon-γ induces expression of MHC class II on intestinal epithelial cells and protects mice from colitis. PLoS ONE 2014, 9, e86844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, A.M.; Kingsbury, D.D.; Wangdi, T.; Xavier, M.N.; Tsolis, R.M.; Bäumler, A.J. Neutrophils are a source of gamma interferon during acute Salmonella enterica serovar Typhimurium colitis. Infect. Immun. 2014, 82, 1692–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, S.Z.; Matsuoka, K.; Kobayashi, T.; Li, F.; Rubinas, T.; Plevy, S.E. Cutting edge: IFN-γ is a negative regulator of IL-23 in murine macrophages and experimental colitis. J. Immun. 2010, 184, 4069–4073. [Google Scholar] [CrossRef] [Green Version]
- Sands, B.E.; Kaplan, G.G. The role of TNFα in ulcerative colitis. J. Clin. Pharmac. 2007, 47, 930–941. [Google Scholar] [CrossRef]
- Roda, G.; Marocchi, M.; Sartini, A.; Roda, E. Cytokine networks in ulcerative colitis. Ulcers 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Whibley, N.; Gaffen, S.L. Gut-busters: IL-17 ain’t afraid of no IL-23. Immunity 2015, 43, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Murga, C.; Laguinge, L.; Wetzker, R.; Cuadrado, A.; Gutkind, J.S. Activation of Akt/Protein Kinase B by G Protein-coupled Receptors A ROLE FOR α AND βγ SUBUNITS OF HETEROTRIMERIC G PROTEINS ACTING THROUGH PHOSPHATIDYLINOSITOL-3-OH KINASEγ. J. Biolog. Chem. 1998, 273, 19080–19085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.L.; Xu, J.; Zhang, X.H.; Qiu, B.Y.; Peng, L.; Zhang, M.; Gan, H.T. PI3K/Akt signaling pathway is involved in the pathogenesis of ulcerative colitis. Inflamm. Res. 2011, 60, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Setia, S.; Nehru, B.; Sanyal, S.N. Upregulation of MAPK/Erk and PI3K/Akt pathways in ulcerative colitis-associated colon cancer. Biomed. Pharmac. 2014, 68, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Garrett, M.D.; Walton, M.I.; Raynaud, F.; de Bono, J.S.; Workman, P. Targeting the PI3K–AKT–mTOR pathway: Progress, pitfalls, and promises. Curr. Opin. Pharm. 2008, 8, 393–412. [Google Scholar] [CrossRef]
- Tokuhira, N.; Kitagishi, Y.; Suzuki, M.; Minami, A.; Nakanishi, A.; Ono, Y.; Kobayashi, K.; Matsuda, S.; Ogura, Y. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy. Int. J. Mol. Med. 2015, 35, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Verstockt, B.; Smith, K.G.; Lee, J.C. Genome-wide association studies in Crohn’s disease: Past, present and future. Clin. Transl. Immun. 2018, 7, e1001. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Strain | Age | Sex | Phenotype | Number | Description |
|---|---|---|---|---|---|
| C57BL/6J | 8–22 weeks | M/F | Wild type (WT) | 16 | Healthy Control |
| Ccr6−/− | 8–22 weeks | M/F | Targeted knockout | 16 | Negative Control |
| Winnie × Ccr6−/− | 8–22 weeks | M/F | Targeted knockout | 16 | Experimental Model |
| Winnie | 8–22 weeks | M/F | Muc2 mutation | 16 | Positive Control |
| Genotype | Body Weight | Diarrhoea | Bloody Faeces | Rectal Prolapse |
|---|---|---|---|---|
| C57BL/6J (WT) | Normal (25 g) | Absent | Absent | Absent |
| Ccr6−/− | Loss of body weight | Absent | Absent | Absent |
| Winnie × Ccr6−/− | Loss of body weight | Absent | Absent | Absent |
| Winnie | Loss of body weight | Present | Absent | Present |
| Genotype | Observation |
|---|---|
| C57BL/6J (WT) | Faecal pellets were well-formed, solid and hard with no symptoms of inflammation, narrow short colon with thin bowel wall (Figure 2A,E) |
| Ccr6−/− | Semi-solid faeces in the proximal colon, faecal pellets formed in the distal colon, mild inflammation, slightly oedematous bowel wall (Figure 2B,F) |
| Winnie × Ccr6−/− | Semi-solid faeces in the proximal colon, faecal pellets formed in distal colon, mild inflammation, mildly thickened bowel wall (Figure 2C,G) |
| Winnie | Watery stools, faecal pellets not formed, high inflammation, thickened bowel wall, reddened and oedematous bowel wall (Figure 2D,H) |
| TH1 | TH2 | TH17 | Treg |
|---|---|---|---|
| IFN-γ | IL-4 | IL-17A | IL-10 |
| TNF-α | IL-6 | IL-23R | |
| IL-18 | IL-13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranasinghe, R.; Fernando, R.; Perera, A.P.; Shastri, M.; Basheer, W.; Scowen, P.; Pinfold, T.; Eri, R. Ccr6 Deficiency Attenuates Spontaneous Chronic Colitis in Winnie. Gastrointest. Disord. 2020, 2, 27-47. https://doi.org/10.3390/gidisord2010004
Ranasinghe R, Fernando R, Perera AP, Shastri M, Basheer W, Scowen P, Pinfold T, Eri R. Ccr6 Deficiency Attenuates Spontaneous Chronic Colitis in Winnie. Gastrointestinal Disorders. 2020; 2(1):27-47. https://doi.org/10.3390/gidisord2010004
Chicago/Turabian StyleRanasinghe, Ranmali, Ruchira Fernando, Agampodi Promoda Perera, Madhur Shastri, Waheedha Basheer, Paul Scowen, Terry Pinfold, and Rajaraman Eri. 2020. "Ccr6 Deficiency Attenuates Spontaneous Chronic Colitis in Winnie" Gastrointestinal Disorders 2, no. 1: 27-47. https://doi.org/10.3390/gidisord2010004
APA StyleRanasinghe, R., Fernando, R., Perera, A. P., Shastri, M., Basheer, W., Scowen, P., Pinfold, T., & Eri, R. (2020). Ccr6 Deficiency Attenuates Spontaneous Chronic Colitis in Winnie. Gastrointestinal Disorders, 2(1), 27-47. https://doi.org/10.3390/gidisord2010004