Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms


{kind=link}
{kind=link}
Abstract
:1. Introduction
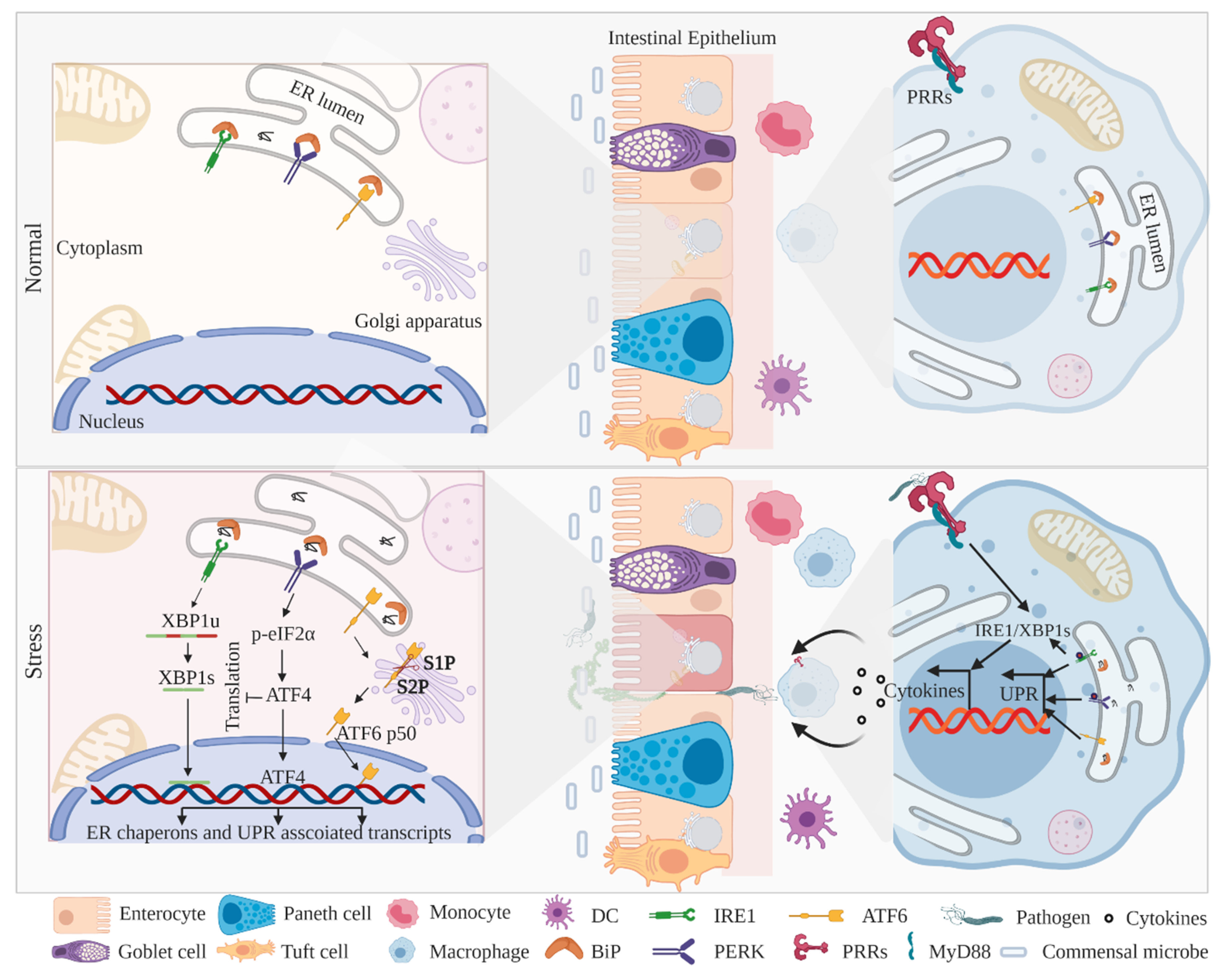
2. ER-Associated UPR Activation and IBD
2.1. IRE1–XBP1 Signaling in IECs and Myeloid Cells
2.2. PERK–eIF2α–CHOP Signaling in IECs and Myeloid Cells
2.3. ATF6 Signaling in IECs and Myeloid Cells
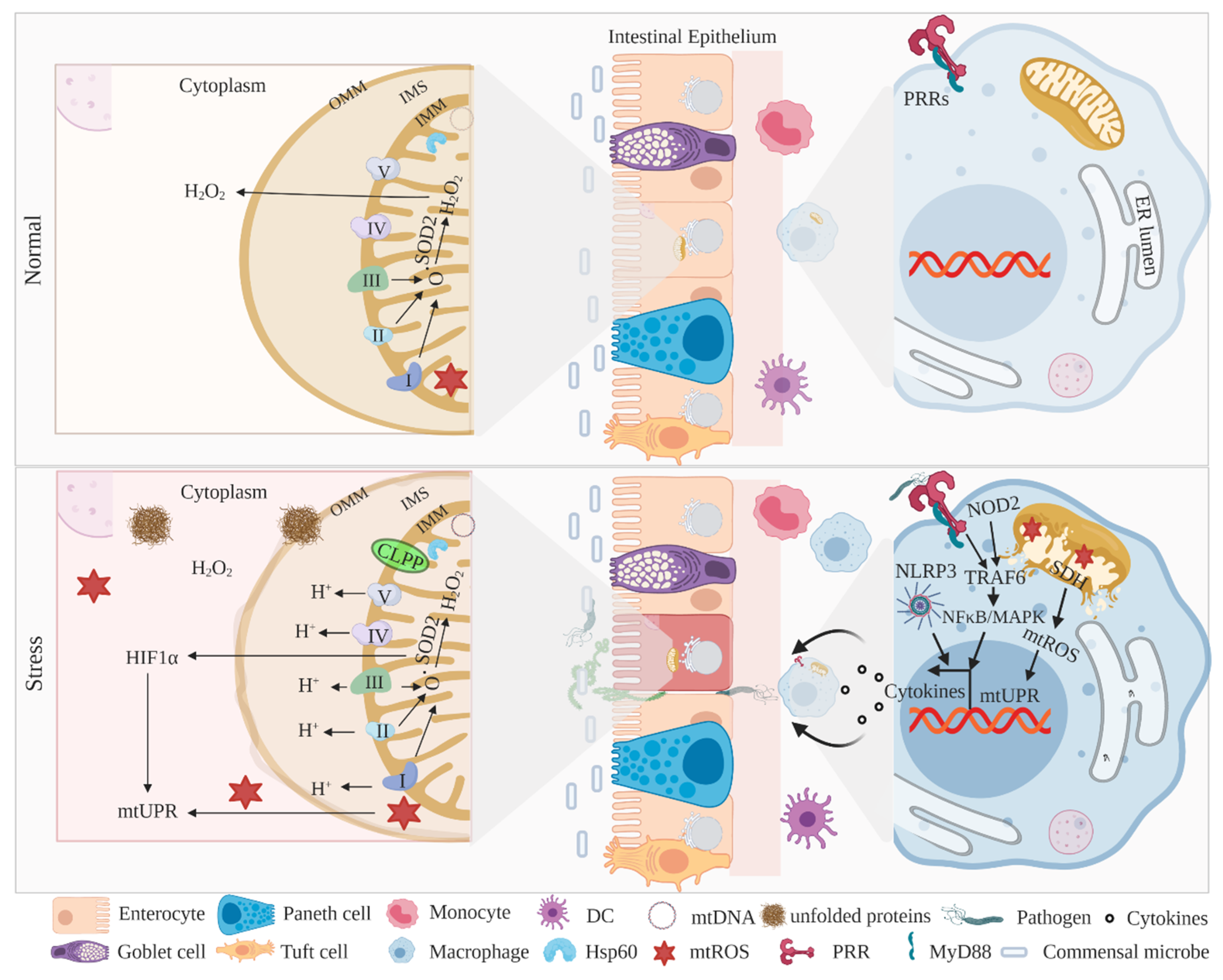
3. Mitochondria-Associated UPR Activation and IBD
3.1. MitoUPR Signaling in IECs and Myeloid Cells
3.2. Mitochondria in the Pathogenesis of IBD
4. Therapeutic Implications Targeting UPR Restoration
5. Conclusion and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Mowat, A.; Agace, W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Ji, M.; Liang, H.-E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 2015, 529, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchenough, G.M.; Johansson, M.E.V.; Gustafsson, J.K.; Bergström, J.H.; Hansson, G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoop, K.A.; Newberry, R.D. Goblet cells: Multifaceted players in immunity at mucosal surfaces. Mucosal Immunol. 2018, 11, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Genet. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Van Der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That MUC2 Is Critical for Colonic Protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef]
- Birchenough, G.M.H.; Nyström, E.E.L.; Johansson, M.E.V.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Oshima, S.; Nakamura, T.; Namiki, S.; Okada, E.; Tsuchiya, K.; Okamoto, R.; Yamazaki, M.; Yokota, T.; Aida, M.; Yamaguchi, Y.; et al. Interferon Regulatory Factor 1 (IRF-1) and IRF-2 Distinctively Up-Regulate Gene Expression and Production of Interleukin-7 in Human Intestinal Epithelial Cells. Mol. Cell. Biol. 2004, 24, 6298–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunliffe, R.N.; Rose, F.R.A.J.; Keyte, J.; Abberley, L.; Chan, W.; Mahida, Y.R. Human defensin 5 is stored in precursor form in normal Paneth cells and is expressed by some villous epithelial cells and by metaplastic Paneth cells in the colon in inflammatory bowel disease. Gut 2001, 48, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevalainen, T.J.; Grönroos, J.M.; Kallajoki, M. Expression of group II phospholipase A2 in the human gastrointestinal tract. Lab. Investig. 1995, 72, 201–208. [Google Scholar] [PubMed]
- Hooper, L.V.; Stappenbeck, T.S.; Hong, C.V.; Gordon, J.I. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003, 4, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin regiiigamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Bevins, C.L. Paneth Cells: Maestros of the Small Intestinal Crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef]
- Ganz, T. Paneth cells—Guardians of the gut cell hatchery. Nat. Immunol. 2000, 1, 99–100. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; Van De Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; Van Royen, M.E.; Van De Werken, H.J.; et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep. 2018, 24, 2312–2328.e7. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Everard, A.; Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 2013, 13, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Van Der Flier, L.G.; Clevers, H. Stem Cells, Self-Renewal, and Differentiation in the Intestinal Epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Van Der Flier, L.G.; Sato, T.; Van Es, J.H.; Born, M.V.D.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; Van Rheenen, J.; Simons, B.D.; et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Born, M.V.D.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2015, 51, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Johansson, M.E.V.; Schütte, A.; Hansson, G.C.; Sjövall, H. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am. J. Physiol. Liver Physiol. 2012, 302, G430–G438. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.V.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2013, 63, 281–291. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J. IL-23 and Autoimmunity: New Insights into the Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Med. 2009, 60, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Barmada, M.M.; Brant, S.R.; Nicolae, D.L.; Achkar, J.-P.; Panhuysen, C.I.; Bayless, T.M.; Cho, J.; Duerr, R.H. A genome scan in 260 inflammatory bowel disease-affected relative pairs. Inflamm. Bowel Dis. 2004, 10, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; The NIDDK IBD Genetics Consortium; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, A.; And the IBSEN Study Group; Balschun, T.; Sina, C.; Ellinghaus, E.; Häsler, R.; Mayr, G.; Albrecht, M.; Wittig, M.; Buchert, E.; et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat. Genet. 2010, 42, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2006, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; Devoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- van der Sloot, K.W.J.; Amini, M.; Peters, V.; Dijkstra, G.; Alizadeh, B.Z. Inflammatory bowel diseases: Review of known environmental protective and risk factors involved. Inflamm. Bowel Dis. 2017, 23, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Adolph, T.E.; Blumberg, R.S. The unfolded protein response and gastrointestinal disease. Semin. Immunopathol. 2013, 35, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Martinez-Naves, E.; Blumberg, R.S. Endoplasmic reticulum stress: Implications for inflammatory bowel disease pathogenesis. Curr. Opin. Gastroenterol. 2010, 26, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, E.; Hironiwa, N.; Kitagawa, M.; Futamura, Y.; Suzuki, S.-I.; Nishio, M.; Imoto, M. ChemInform Abstract: Trierixin, a Novel Inhibitor of ER Stress-Induced XBP1 Activation from Streptomyces sp. Part 1. Taxonomy, Fermentation, Isolation, and Biological Activities. ChemInfrom 2008, 39, 547–553. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kaplan, G.G. Environmental Risk Factors for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2010, 6, 339–346. [Google Scholar]
- Rogler, G.; Zeitz, J.; Biedermann, L. The Search for Causative Environmental Factors in Inflammatory Bowel Disease. Dig. Dis. 2016, 34, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosnes, J. What is the link between the use of tobacco and ibd? Inflamm. Bowel Dis. 2008, 14, S14–S15. [Google Scholar] [CrossRef] [PubMed]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and Natural History of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1785–1794.e4. [Google Scholar] [CrossRef]
- Birrenbach, T.; Böcker, U. Inflammatory bowel disease and smoking: A review of epidemiology, pathophysiology, and therapeutic implications. Inflamm. Bowel Dis. 2004, 10, 848–859. [Google Scholar] [CrossRef]
- Verschuere, S.; Bracke, K.R.; Demoor, T.; Plantinga, M.; Verbrugghe, P.L.; Ferdinande, L.; Lambrecht, B.N.; Brusselle, G.G.G.; Cuvelier, C.A. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab. Investig. 2011, 91, 1056–1067. [Google Scholar] [CrossRef]
- Berg, D.J.; Zhang, J.; Weinstock, J.V.; Ismail, H.F.; Earle, K.A.; Alila, H.; Pamukcu, R.; Moore, S.A.; Lynch, R.G. Rapid development of colitis in NSAID-treated IL-10–deficient mice. Gastroenterology 2002, 123, 1527–1542. [Google Scholar] [CrossRef]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2014, 64, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Author Correction: Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 898. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; MacPherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Medica 2019, 110, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Rosso, C.; Ribaldone, D.G.; Dughera, F.; Fagoonee, S.; Astegiano, M.; Pellicano, R. Physiopathology of intestinal barrier and the role of zonulin. Minerva Biotecnol. 2019, 31, 83–92. [Google Scholar] [CrossRef]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Chen, Y.; Fan, L.; Xi, Q.-L.; Wu, G.-H.; Li, X.-X.; Yuan, T.-L.; He, S.-Q.; Yu, Y.; Shao, M.; et al. The Endoplasmic Reticulum Stress Sensor IRE1α in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J. Biol. Chem. 2015, 290, 15327–15336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.-M.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Broadley, S.A.; Hartl, F.U. Mitochondrial stress signaling: A pathway unfolds. Trends Cell Biol. 2008, 18, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—Gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2011, 61, 1269–1278. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.D.; Price, G.R.; Tauro, S.B.; Taupin, U.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant Mucin Assembly in Mice Causes Endoplasmic Reticulum Stress and Spontaneous Inflammation Resembling Ulcerative Colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in ire1beta-deficient mice. J. Clin. Investg. 2001, 107, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Bakunts, A.; Orsi, A.; Vitale, M.; Cattaneo, A.; Lari, F.; Tadè, L.; Sitia, R.; Raimondi, A.; Bachi, A.; Van Anken, E. Ratiometric sensing of BiP-client versus BiP levels by the unfolded protein response determines its signaling amplitude. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.-Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic ifn-beta induction via x-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Núñez, G.; Grethe, J.S.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Hedl, M.; Ranjan, K.; Abraham, C. LACC1 Required for NOD2-Induced, ER Stress-Mediated Innate Immune Outcomes in Human Macrophages and LACC1 Risk Variants Modulate These Outcomes. Cell Rep. 2019, 29, 4525–4539.e4. [Google Scholar] [CrossRef] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Kopp, M.C.; Nowak, P.R.; Larburu, N.; Adams, C.J.; Ali, M.M.U. In vitro FRET analysis of IRE1 and BiP association and dissociation upon endoplasmic reticulum stress. eLife 2018, 7, e30257. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. Ire1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, B.; Iwakoshi, N.N.; Glimcher, L.H.; Ploegh, H.L. Rapid Turnover of Unspliced Xbp-1 as a Factor That Modulates the Unfolded Protein Response. J. Biol. Chem. 2005, 281, 5852–5860. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nature 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M. The ER stress transducer ire1beta is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, Y.; Hosoda, A.; Sasaka, S.-I.; Tsuru, A.; Kohno, K. RNase domains determine the functional difference between IRE1α and IRE1β. FEBS Lett. 2008, 582, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Grey, M.J.; Cloots, E.; Simpson, M.S.; LeDuc, N.; Serebrenik, Y.V.; De Luca, H.; De Sutter, D.; Luong, P.; Thiagarajah, J.R.; Paton, A.W.; et al. IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. J. Cell Biol. 2020, 219, e201904048. [Google Scholar] [CrossRef] [Green Version]
- Tsuru, A.; Fujimoto, N.; Takahashi, S.; Saito, M.; Nakamura, D.; Iwano, M.; Iwawaki, T.; Kadokura, H.; Ron, D.; Kohno, K. Negative feedback by ire1beta optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2864–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillich, H.; Loose, M.; Zimmer, K.-P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Kim, M.-J.; Blumenthal, A.; Koo, J.; Ehrt, S.; Wainwright, H.; Bekker, L.-G.; Kaplan, G.; Nathan, C.; Tabas, I.; et al. Induction of ER Stress in Macrophages of Tuberculosis Granulomas. PLoS ONE 2010, 5, e12772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.S.; Jeong, S.O.; Pae, H.O.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Endoplasmic reticulum stress-induced ire1alpha activation mediates cross-talk of gsk-3beta and xbp-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, S.; Rutgeerts, P.; Van Steen, K.; Joossens, S.; Claessens, G.; Pierik, M.; Peeters, M.; Vlietinck, R. Genome wide scan in a Flemish inflammatory bowel disease population: Support for the IBD4 locus, population heterogeneity, and epistasis. Gut 2004, 53, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Hampe, J.; Schreiber, S.; Shaw, S.H.; Lau, K.F.; Bridger, S.; MacPherson, A.J.; Cardon, L.R.; Sakul, H.; Harris, T.J.; Buckler, A.; et al. A Genomewide Analysis Provides Evidence for Novel Linkages in Inflammatory Bowel Disease in a Large European Cohort. Am. J. Hum. Genet. 1999, 64, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M.U. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Cao, S.S.; Harrington, J.C.; Chuang, B.-M.; Kaufman, R.J.; Wang, M.; Eckmann, L. Phosphorylation of eIF2α Is Dispensable for Differentiation but Required at a Posttranscriptional Level for Paneth Cell Function and Intestinal Homeostasis in Mice. Inflamm. Bowel Dis. 2014, 20, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000.e6. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Namba, T.; Tanaka, K.-I.; Ito, Y.; Ishihara, T.; Hoshino, T.; Gotoh, T.; Endo, M.; Sato, K.; Mizushima, T. Positive Role of CCAAT/Enhancer-Binding Protein Homologous Protein, a Transcription Factor Involved in the Endoplasmic Reticulum Stress Response in the Development of Colitis. Am. J. Pathol. 2009, 174, 1786–1798. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S.H. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.W.; Cui, D.; Arellano, J.; Dorweiler, B.; Harding, H.P.; Fitzgerald, K.A.; Ron, D.; Tabas, I. Adaptive suppression of the ATF4–CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 2009, 11, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Shkoda, A.; Ruiz, P.A.; Daniel, H.; Kim, S.C.; Rogler, G.; Sartor, R.B.; Haller, D. Interleukin-10 Blocked Endoplasmic Reticulum Stress in Intestinal Epithelial Cells: Impact on Chronic Inflammation. Gastroenterology 2007, 132, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Peterson, R.L.; Twine, N.C.; Zuberek, K.A.; Brodeur, B.J.; Casciotti, L.; Maganti, V.; Reddy, P.S.; Strahs, A.; Immermann, F.; et al. Molecular Classification of Crohn’s Disease and Ulcerative Colitis Patients Using Transcriptional Profiles in Peripheral Blood Mononuclear Cells. J. Mol. Diagn. 2006, 8, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaert, S.; De Vos, M.; Olievier, K.; Peeters, H.; Elewaut, D.; Lambrecht, B.; Pouliot, P.; Laukens, D. Involvement of Endoplasmic Reticulum Stress in Inflammatory Bowel Disease: A Different Implication for Colonic and Ileal Disease? PLoS ONE 2011, 6, e25589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treton, X.; Pedruzzi, E.; Cazals–Hatem, D.; Grodet, A.; Panis, Y.; Groyer, A.; Moreau, R.; Bouhnik, Y.; Daniel, F.; Ogier-Denis, E. Altered Endoplasmic Reticulum Stress Affects Translation in Inactive Colon Tissue From Patients With Ulcerative Colitis. Gastroenterology 2011, 141, 1024–1035. [Google Scholar] [CrossRef]
- Hu, X.; Deng, J.; Yu, T.; Chen, S.; Ge, Y.; Zhou, Z.; Guo, Y.; Ying, H.; Zhai, Q.; Chen, Y.; et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019, 156, 1098–1111. [Google Scholar] [CrossRef]
- Jorgensen, E.D.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 2008, 45, 50–59. [Google Scholar] [CrossRef]
- Adair-Kirk, T.L.; Atkinson, J.J.; Griffin, G.L.; Watson, M.A.; Kelley, D.G.; Demello, D.; Senior, R.M.; Betsuyaku, T. Distal Airways in Mice Exposed to Cigarette Smoke: Nrf2-regulated genes are increased in clara cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Hackett, N.R.; Heguy, A.; Harvey, B.-G.; O’Connor, T.P.; Luettich, K.; Flieder, D.B.; Kaplan, R.; Crystal, R.G. Variability of Antioxidant-Related Gene Expression in the Airway Epithelium of Cigarette Smokers. Am. J. Respir. Cell Mol. Biol. 2003, 29, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.; O’Hare, P. Transmembrane bZIP Transcription Factors in ER Stress Signaling and the Unfolded Protein Response. Antioxid. Redox Signal. 2007, 9, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable Binding of ATF6 to BiP in the Endoplasmic Reticulum Stress Response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandl, K.; Rutschmann, S.; Li, X.; Du, X.; Xiao, N.; Schnabl, B.; Brenner, D.A.; Beutler, B. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc. Natl. Acad. Sci. USA 2009, 106, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Asada, R.; Saito, A.; Kawasaki, N.; Kanemoto, S.; Iwamoto, H.; Oki, M.; Miyagi, H.; Izumi, S.; Imaizumi, K. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J. Biol. Chem. 2012, 287, 8144–8153. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Saito, A.; Asada, R.; Kanemoto, S.; Imaizumi, K. Increased Susceptibility to Dextran Sulfate Sodium-Induced Colitis in the Endoplasmic Reticulum Stress Transducer OASIS Deficient Mice. PLoS ONE 2014, 9, e88048. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C.; Zhu, H.; Toso, R.; Camire, R.M. Effects of the Isoform-specific Characteristics of ATF6 α and ATF6 β on Endoplasmic Reticulum Stress Response Gene Expression and Cell Viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, D.J.; Morrison, L.; Glembotski, C.C. Opposing roles for atf6alpha and atf6beta in endoplasmic reticulum stress response gene induction. J. Biol. Chem. 2004, 279, 21078–21084. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 Mediates a Pro-Inflammatory Synergy Between ER Stress and TLR Activation in the Pathogenesis of Liver Ischemia-Reperfusion Injury. Arab. Archaeol. Epigr. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic Reticulum Stress Promotes Macrophage-derived Foam Cell Formation by Up-regulating Cluster of Differentiation 36 (CD36) Expression. J. Biol. Chem. 2013, 289, 4032–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, K.; Pathak, C. Expression of FADD and cFLIPL balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol. Cell. Biochem. 2016, 422, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.-M.; Rungi, A.A.; Robinson, B.H.; Hood, D.A. Compensatory responses of protein import and transcription factor expression in mitochondrial DNA defects. Am. J. Physiol. Physiol. Cell 2004, 286, C867–C875. [Google Scholar] [CrossRef]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (mtUPR) and Cognate Promoter Elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-Nuclear Communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef]
- Fung, K.Y.; Brierley, G.V.; Henderson, S.; Hoffmann, P.; McColl, S.R.; Lockett, T.J.; Head, R.; Cosgrove, L. Butyrate-Induced Apoptosis in HCT116 Colorectal Cancer Cells Includes Induction of a Cell Stress Response. J. Proteome Res. 2011, 10, 1860–1869. [Google Scholar] [CrossRef]
- Kolar, S.; Barhoumi, R.; Jones, C.K.; Wesley, J.; Lupton, J.R.; Fan, Y.-Y.; Chapkin, R.S.; Bs, C.K.J. Interactive effects of fatty acid and butyrate-induced mitochondrial Ca2+ loading and apoptosis in colonocytes. Cancer 2011, 117, 5294–5303. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Hart, P.; Germain, D.; Bonini, M.G. SOD2 and the Mitochondrial UPR: Partners Regulating Cellular Phenotypic Transitions. Trends Biochem. Sci. 2016, 41, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Rath, E.; Haller, D. Mitochondria at the Interface Between Danger Signaling and Metabolism: Role of Unfolded Protein Responses in Chronic Inflammation. Inflamm. Bowel Dis. 2012, 18, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; Van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Schürmann, G.; Brüwer, M.; Klotz, A.; Schmid, K.W.; Senninger, N.; Zimmer, K.-P. Transepithelial transport processes at the intestinal mucosa in inflammatory bowel disease. Int. J. Color. Dis. 1999, 14, 41–46. [Google Scholar] [CrossRef]
- Heller, S.; Penrose, H.M.; Cable, C.; Biswas, D.; Nakhoul, H.; Baddoo, M.; Flemington, E.; Crawford, S.E.; Savkovic, S.D. Reduced mitochondrial activity in colonocytes facilitates ampkalpha2-dependent inflammation. FASEB J. 2017, 31, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzen, L.E.; Lindmark, T.; Wiren, M.; Tagesson, C.; Sjodahl, R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of crohn’s disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.T.; Aird, R.E.; Liu, B.; Boyapati, R.K.; Kennedy, N.A.; Dorward, D.A.; Noble, C.L.; Shimizu, T.; Carter, R.N.; Chew, E.T.; et al. MDR1-deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 2017, 11, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Waldschmitt, N.; Berger, E.; Rath, E.; Sartor, R.B.; Weigmann, B.; Heikenwälder, M.; Gerhard, M.; Janssen, K.-P.; Haller, D. C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol. 2014, 7, 1452–1466. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The Chop Gene Contains an Element for the Positive Regulation of the Mitochondrial Unfolded Protein Response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Papazoglou, D.; Simopoulos, C.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Hypoxia inducible factor 1α and 2α overexpression in inflammatory bowel disease. J. Clin. Pathol. 2003, 56, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitouni, N.E.; Chotikatum, S.; Von Köckritz-Blickwede, M.; Naim, H.Y. The impact of hypoxia on intestinal epithelial cell functions: Consequences for invasion by bacterial pathogens. Mol. Cell. Pediatr. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Theiss, A.L.; Idell, R.D.; Srinivasan, S.; Klapproth, J.-M.; Jones, D.P.; Merlin, D.; Sitaraman, S.V. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2006, 21, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; De Jong, L.; Artal-Sanz, M.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; Van Der Spek, H.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J.C. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002, 166, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Próchnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (dss) is mediated by the nlrp3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wi, S.M.; Moon, G.; Kim, J.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. Tak1-ecsit-traf6 complex plays a key role in the tlr4 signal to activate nf-kappab. J. Biol. Chem. 2014, 289, 35205–35214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Lahiri, A.; Hedl, M.; Yan, J.; Abraham, C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nat. Commun. 2017, 8, 15614. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W. The colonic epithelium in ulcerative colitis: An energy-deficiency disease? Lancet 1980, 316, 712–715. [Google Scholar] [CrossRef]
- Dankowski, T.; Schröder, T.; Möller, S.; Yu, X.; Ellinghaus, D.; Bär, F.; Fellermann, K.; Lehnert, H.; Stefansson, K.; Franke, A.; et al. Male-specific association between MT-ND4 11719 A/G polymorphism and ulcerative colitis: A mitochondria-wide genetic association study. BMC Gastroenterol. 2016, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Abrantes, P.; Sousa, I.; Francisco, V.; Santos, P.; Francisco, D.; Xavier, J.M.; Oliveira, S.A. Ulcerative Colitis Is Under Dual (Mitochondrial and Nuclear) Genetic Control. Inflamm. Bowel Dis. 2016, 22, 774–781. [Google Scholar] [CrossRef]
- Lin, Z.; Nelson, L.; Franke, A.; Poritz, L.; Li, T.-Y.; Wu, R.; Wang, Y.; MacNeill, C.; Thomas, N.J.; Stefansson, K.; et al. OCTN1 variant L503F is associated with familial and sporadic inflammatory bowel disease. J. Crohns Coliti 2010, 4, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, S.; Tremelling, M.; Bredin, F.; Godfrey, L.; Howson, J.M.M.; Parkes, M. Evidence for association of OCTN genes and IBD5 with ulcerative colitis. Gut 2006, 55, 809–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekhawat, P.S.; Srinivas, S.R.; Matern, D.; Bennett, M.J.; Boriack, R.; George, V.; Xu, H.; Prasad, P.D.; Roon, P.; Ganapathy, V. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (octn2(-/-)) mice. Mol. Genet. Metab. 2007, 92, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef] [PubMed]
- Rogala, A.R.; Schoenborn, A.A.; Fee, B.E.; Cantillana, V.A.; Joyce, M.J.; Gharaibeh, R.Z.; Roy, S.; Fodor, A.A.; Sartor, R.B.; Taylor, G.A.; et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model. Mech. 2017, 11, dmm031070. [Google Scholar] [CrossRef] [Green Version]
- Haberman, Y.; Karns, R.; Dexheimer, P.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Keita, Å.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting Mitochondria-Derived Reactive Oxygen Species to Reduce Epithelial Barrier Dysfunction and Colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef] [Green Version]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2006, 56, 61–72. [Google Scholar] [CrossRef]
- Büning, C.; Geissler, N.; Prager, M.; Sturm, A.; Baumgart, D.C.; Büttner, J.; Bühner, S.; Haas, V.; Lochs, H. Increased small intestinal permeability in ulcerative colitis: Rather genetic than environmental and a risk factor for extensive disease? Inflamm. Bowel Dis. 2012, 18, 1932–1939. [Google Scholar] [CrossRef]
- Hsieh, S.-Y.; Shih, T.-C.; Yeh, C.-Y.; Lin, C.-J.; Chou, Y.-Y.; Lee, Y.-S. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 2006, 6, 5322–5331. [Google Scholar] [CrossRef]
- Pereira, C.C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative stress and DNA damage: Implications in inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, R.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, E.; Haller, D. Structure–function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2011, 409, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; San-Miguel, B.; Prause, C.; Marroni, N.; Cuevas, M.J.; González-Gallego, J.; Tuñón, M.J. Glutamine Treatment Attenuates Endoplasmic Reticulum Stress and Apoptosis in TNBS-Induced Colitis. PLoS ONE 2012, 7, e50407. [Google Scholar] [CrossRef]
- Koh, S.J.; Kim, J.W.; Kim, B.G.; Lee, K.L.; Chun, J.; Kim, J.S. Fexofenadine regulates nuclear factor-kappab signaling and endoplasmic reticulum stress in intestinal epithelial cells and ameliorates acute and chronic colitis in mice. J. Pharmacol. Exp. Ther. 2015, 352, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, N.; Kaufman, R.J.; Ma, D.; Coen, N.M.; Ron, D.; et al. A Selective Inhibitor of eIF2α Dephosphorylation Protects Cells from ER Stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Okazaki, T.; Nishio, A.; Takeo, M.; Sakaguchi, Y.; Fukui, T.; Uchida, K.; Okazaki, K. Inhibition of the Dephosphorylation of Eukaryotic Initiation Factor 2α Ameliorates Murine Experimental Colitis. Digestion 2014, 90, 147–158. [Google Scholar] [CrossRef]
- Rauthan, M.; Pilon, M. A chemical screen to identify inducers of the mitochondrial unfolded protein response in C. elegans. Worm 2015, 4, e1096490. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointest. Disord. 2020, 2, 246-266. https://doi.org/10.3390/gidisord2030024
Ranjan K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointestinal Disorders. 2020; 2(3):246-266. https://doi.org/10.3390/gidisord2030024
Chicago/Turabian StyleRanjan, Kishu. 2020. "Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms" Gastrointestinal Disorders 2, no. 3: 246-266. https://doi.org/10.3390/gidisord2030024
APA StyleRanjan, K. (2020). Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointestinal Disorders, 2(3), 246-266. https://doi.org/10.3390/gidisord2030024