From Frustrated Packing to Tecton-Driven Porous Molecular Solids


Abstract
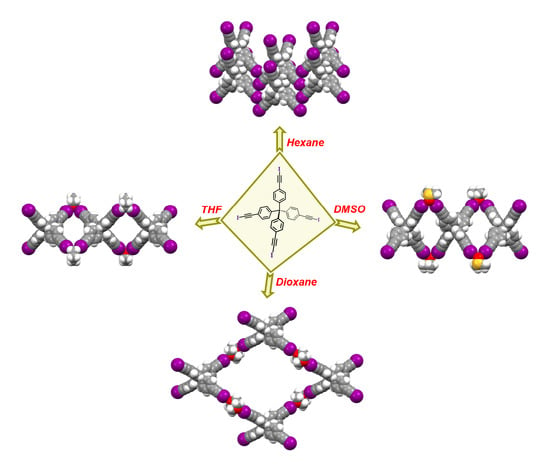
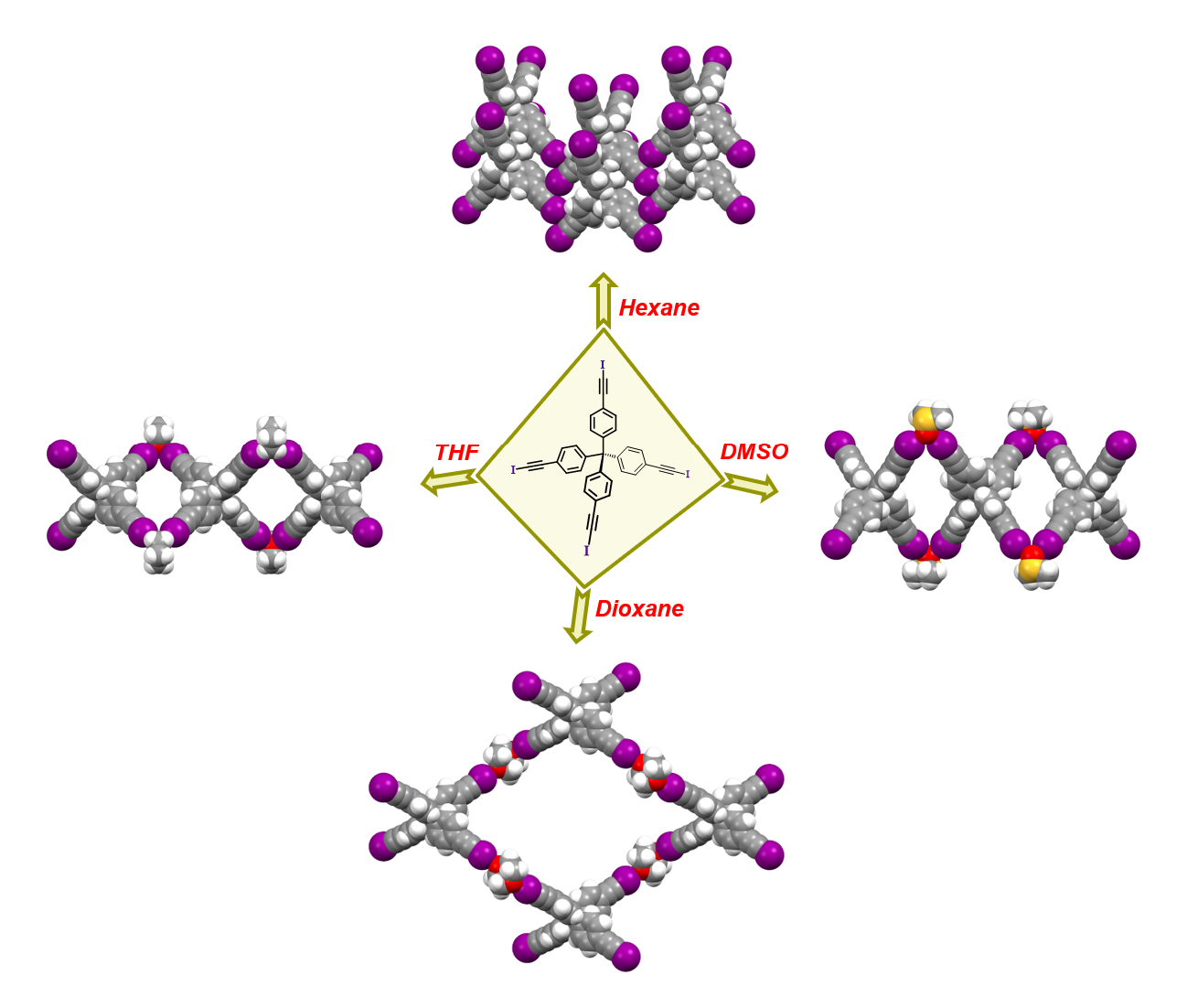
:
1. Introduction
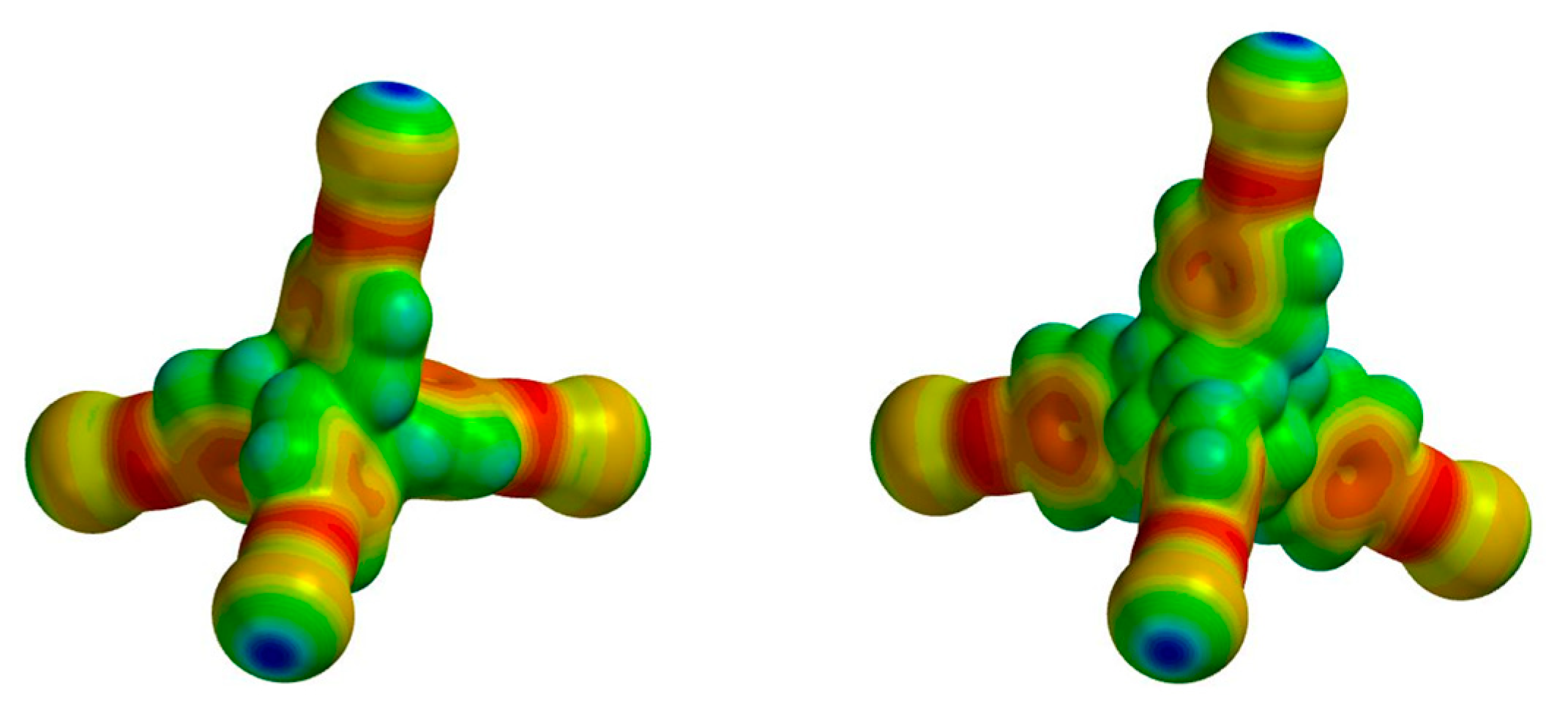
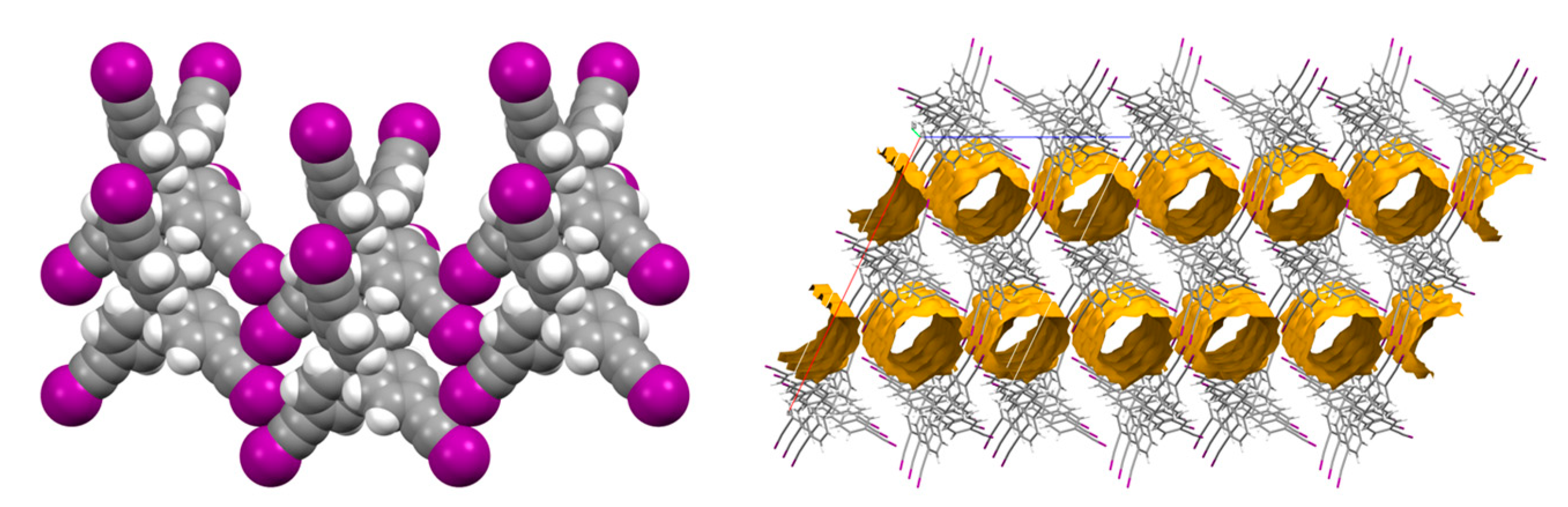
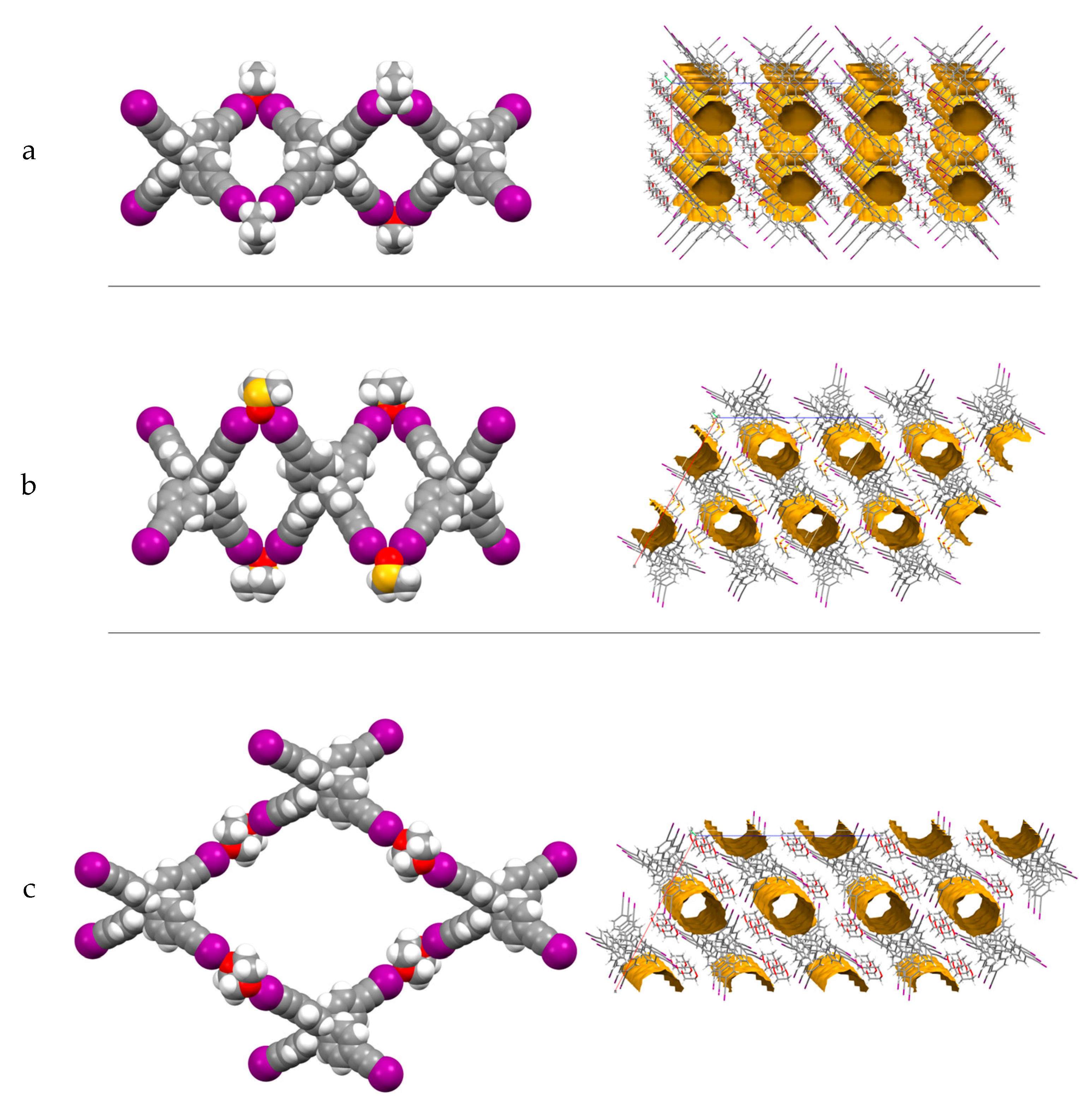
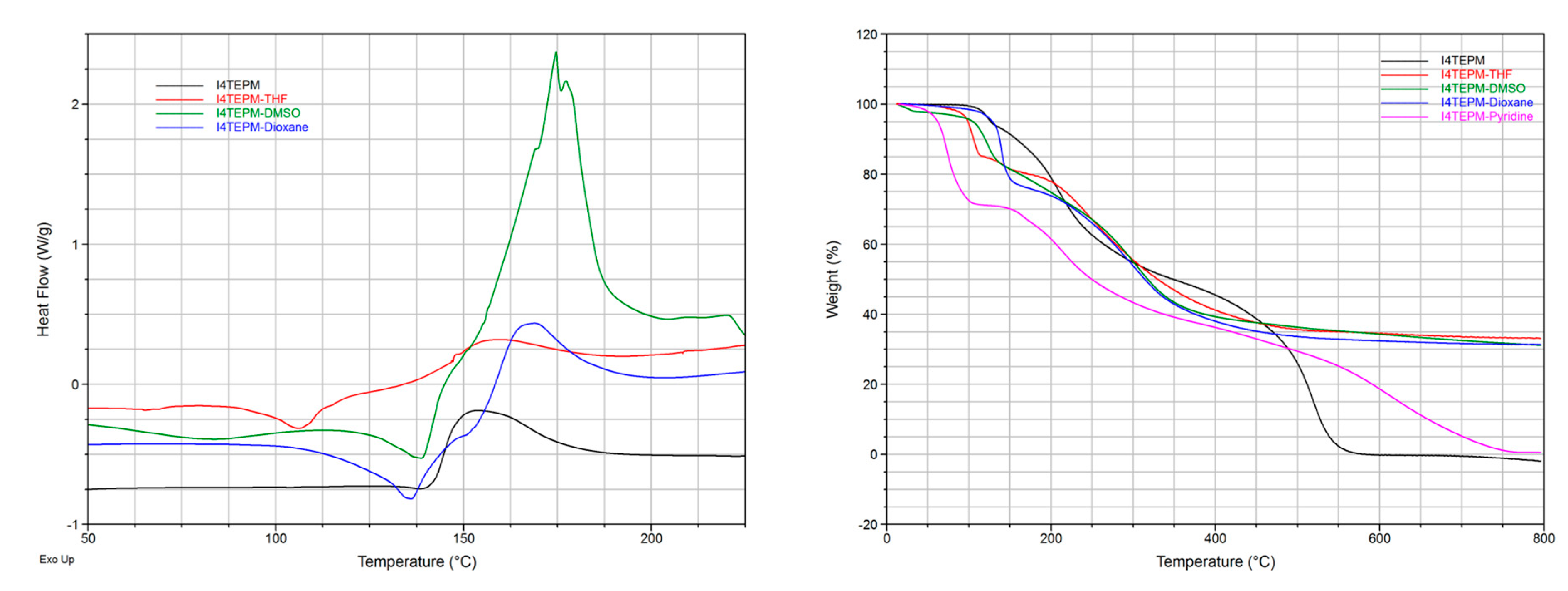
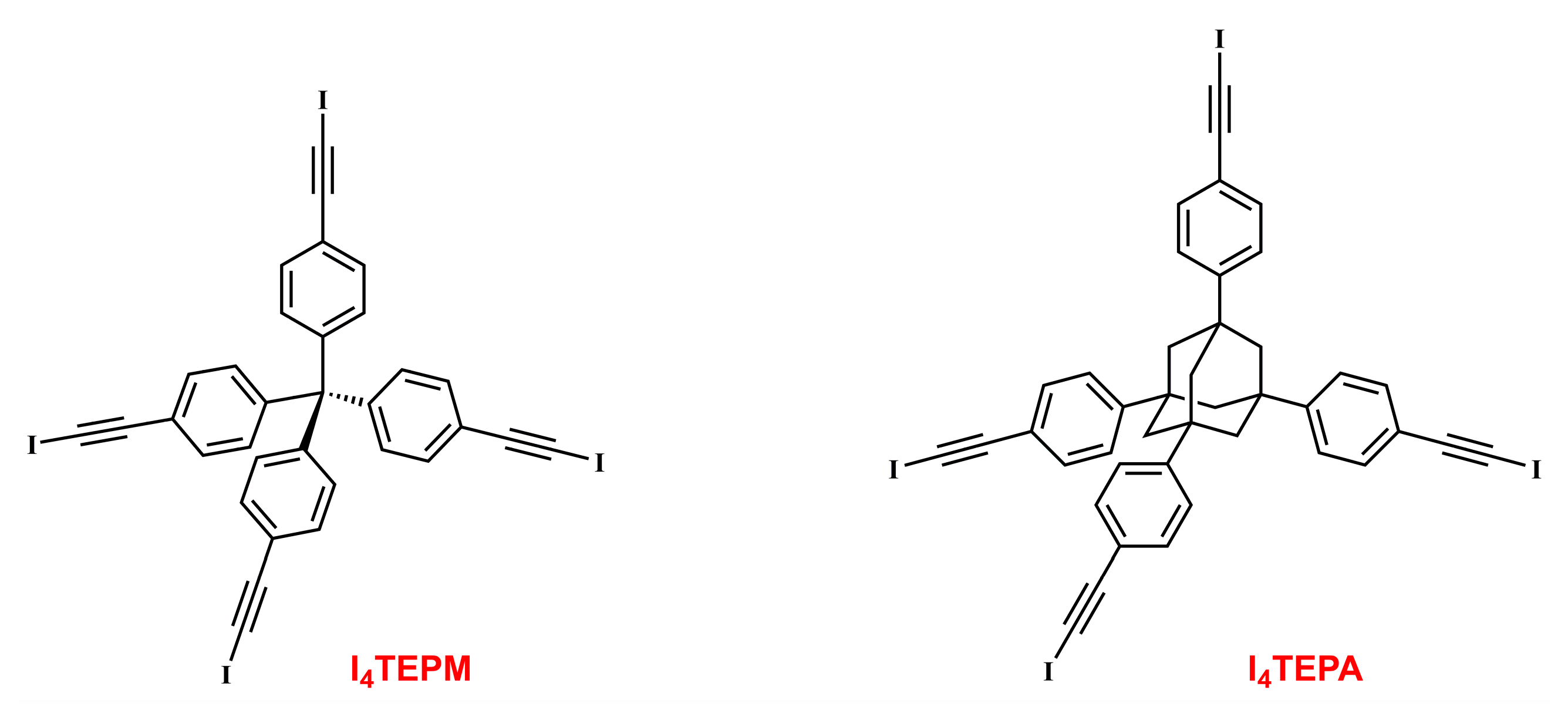
2. Results and Discussion
3. Conclusions
4. Materials and Methods
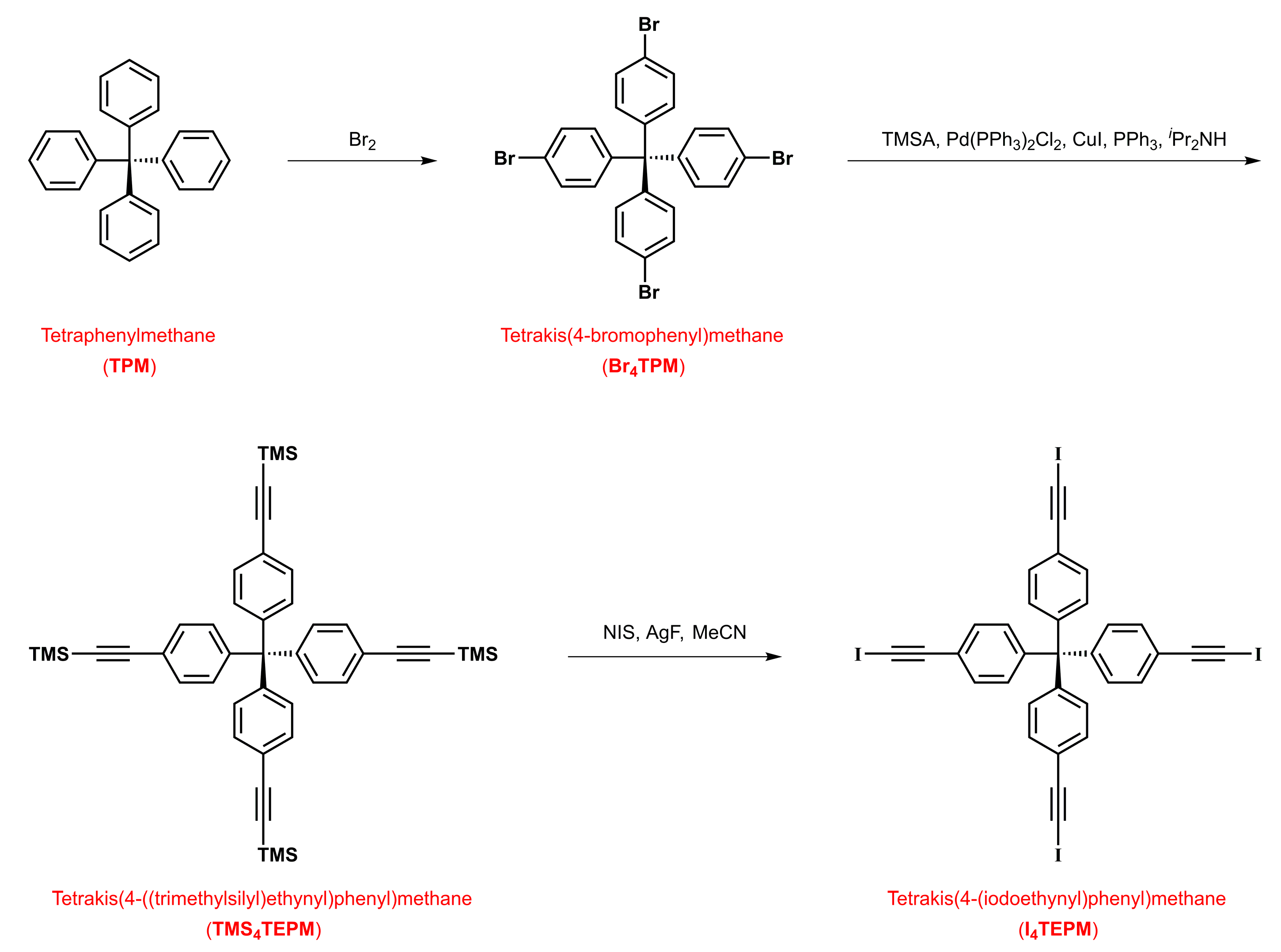
4.1. Synthesis of Tetrakis(4-bromophenyl)methane (Br4TPM)
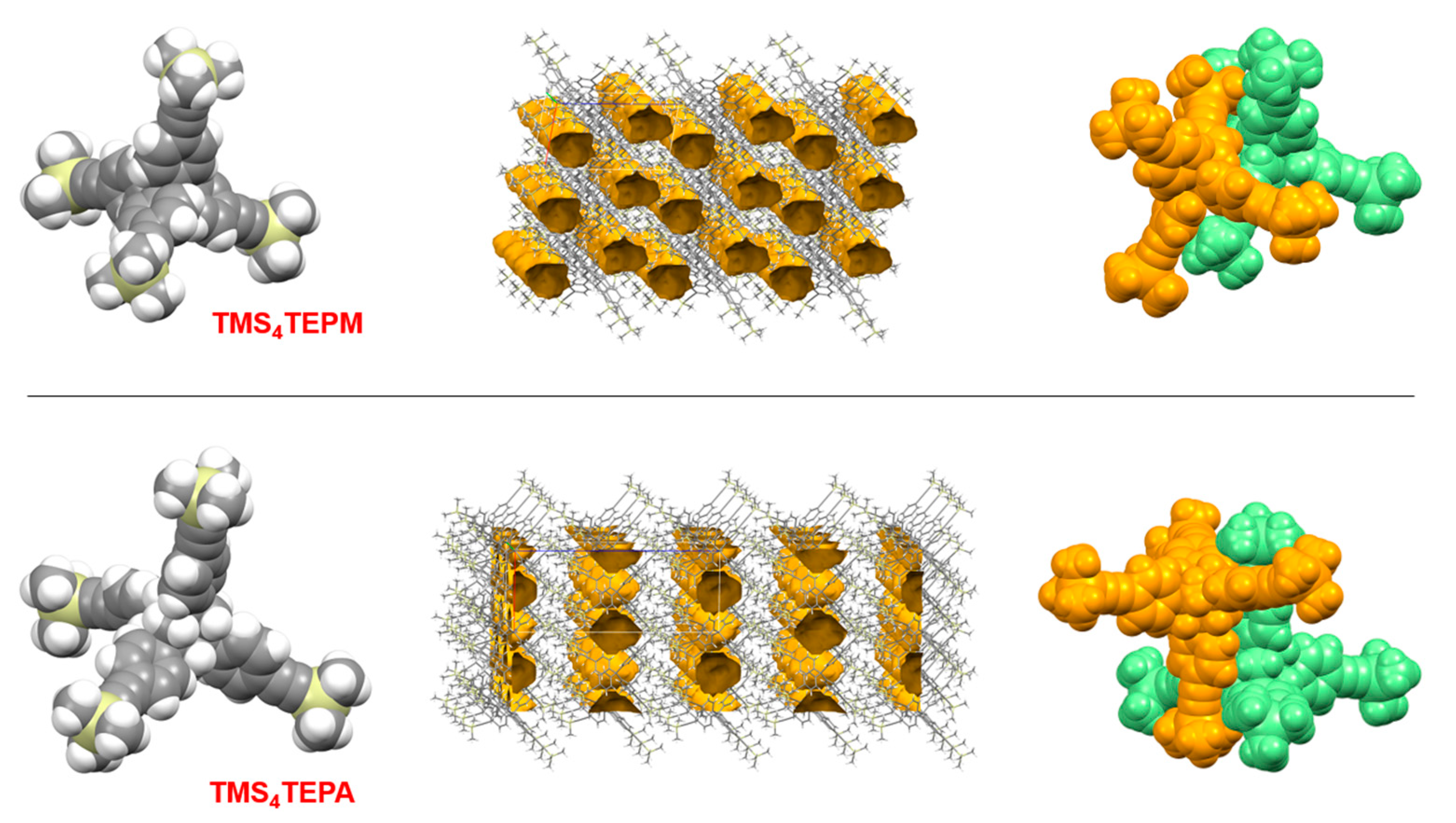
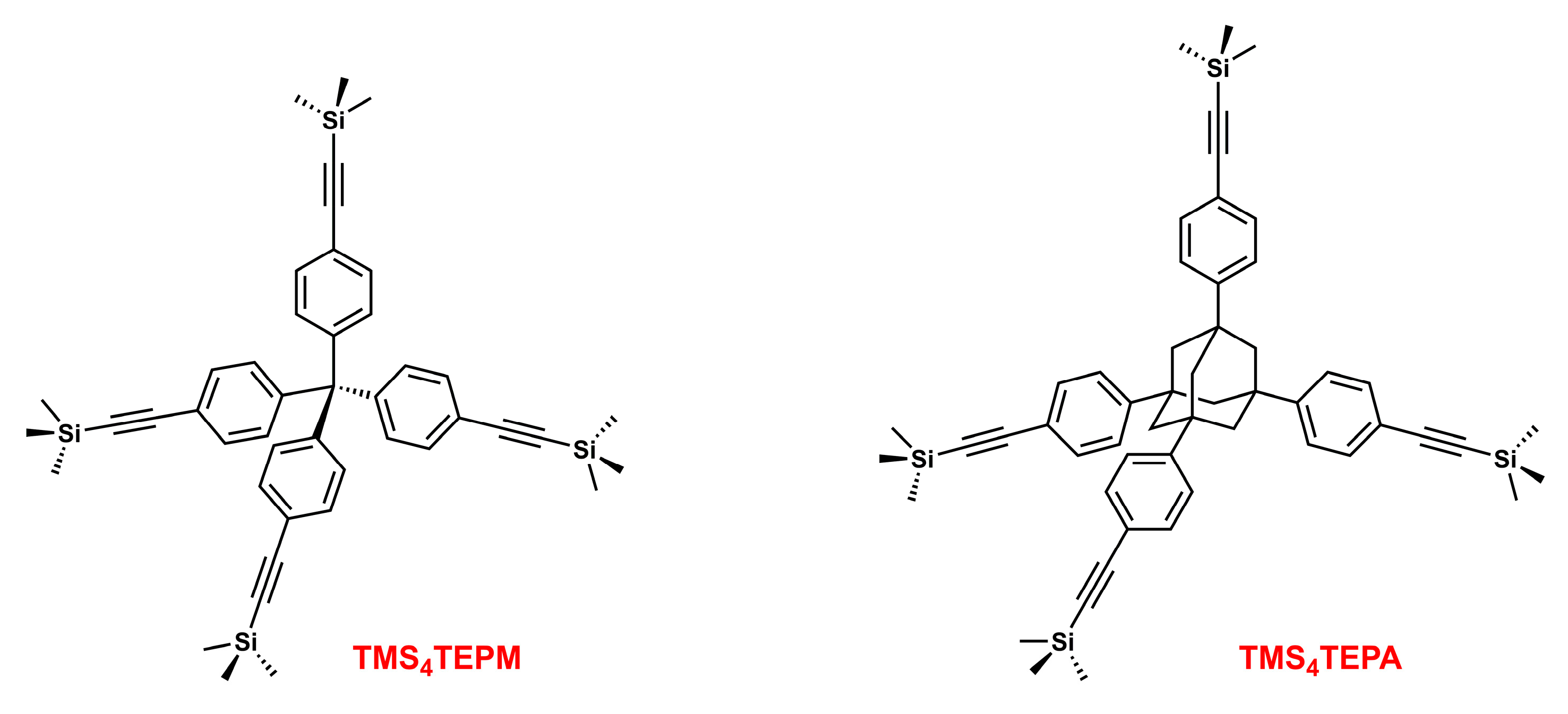
4.2. Synthesis of Tetrakis(4-((trimethylsilyl)ethynyl)phenyl)methane (TMS4TEPM)
4.3. Synthesis of Tetrakis(4-(iodoethynyl)phenyl)methane (I4TEPM)
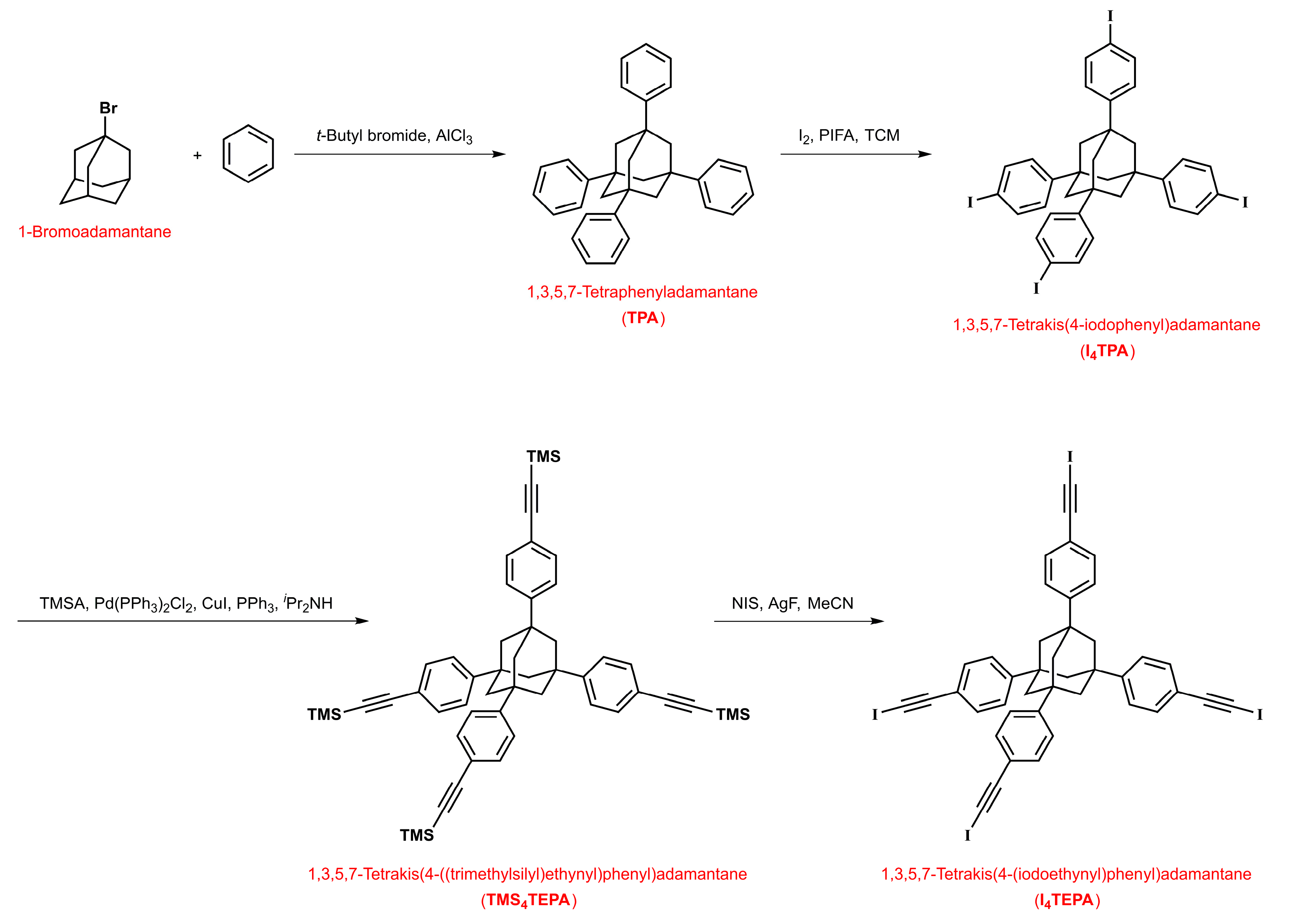
4.4. Synthesis of 1,3,5,7-Tetraphenyladamantane (TPA)
4.5. Synthesis of 1,3,5,7-Tetrakis(4-iodophenyl)adamantane (I4TPA)
4.6. Synthesis of 1,3,5,7-Tetrakis(4-((trimethylsilyl)ethynyl)phenyl)adamantane (TMS4TEPA)
4.7. Synthesis of 1,3,5,7-Tetrakis(4-(iodoethynyl)phenyl)adamantane (I4TEPA)
4.8. Synthesis of I4TEPM·4pyridine
4.9. Synthesis of I4TEPM·2THF
4.10. Synthesis of I4TEPM·2DMSO
4.11. Synthesis of I4TEPM·2dioxane
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kitaigorodskii, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Kitaigorodskii, A.I. Organic Chemical Crystallography; Consultants Bureau: New York, NY, USA, 1961. [Google Scholar]
- Kitaigorodskii, A.I. Non-bonded interactions of atoms in organic crystals and molecules. Chem. Soc. Rev. 1978, 7, 133–163. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.I. The principle of close packing and the condition of thermodynamic stability of organic crystals. Acta Crystallogr. 1965, 18, 585–590. [Google Scholar] [CrossRef]
- Kitaigorodskii, A.I. The close-packing of molecules in crystals of organic compounds. J. Phys. (USSR) 1945, 9, 351–352. [Google Scholar]
- Lü, J.; Cao, R. Porous organic molecular frameworks with extrinsic porosity: A platform for carbon storage and separation. Angew. Chem. Int. Ed. 2016, 55, 9474–9480. [Google Scholar] [CrossRef] [PubMed]
- Hashim, M.I.; Hsu, C.-W.; Le, H.T.M.; Miljanić, O.Š. Organic molecules with porous crystal structures. Synlett 2016, 27, 1907–1918. [Google Scholar]
- Tian, J.; Thallapally, P.K.; McGrail, B.P. Porous organic molecular materials. CrystEngComm 2012, 14, 1909–1919. [Google Scholar] [CrossRef]
- Mastalerz, M. Permanent porous materials from discrete organic molecules—towards ultra-high surface areas. Chem. Eur. J. 2012, 18, 10082–10091. [Google Scholar] [CrossRef]
- McKeown, N.B. Nanoporous molecular crystals. J. Mater. Chem. 2010, 20, 10588–10597. [Google Scholar] [CrossRef] [Green Version]
- Holst, J.R.; Trewin, A.; Cooper, A.I. Porous organic molecules. Nat. Chem. 2010, 2, 915–920. [Google Scholar] [CrossRef]
- Hasell, T.; Cooper, A.I. Porous organic cages: Soluble, modular and molecular pores. Nat. Rev. Mater. 2016, 1, 16053. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D.; Sumby, C.J.; Doonan, C.J. Synthesis and applications of porous organic cages. Chem. Lett. 2015, 44, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Mastalerz, M. Organic cage compounds—from shape-persistency to function. Chem. Soc. Rev. 2014, 43, 1934–1947. [Google Scholar] [CrossRef] [PubMed]
- Imashiro, F.; Yoshimura, M.; Fujiwara, T. ‘Guest-free’ Dianin’s compound. Acta Crystallogr. C 1998, 54, 1357–1360. [Google Scholar] [CrossRef]
- Barrer, R.M.; Shanson, V.H. Dianin’s compound as a zeolitic sorbent. J. Chem. Soc. Chem. Commun. 1976, 333–334. [Google Scholar] [CrossRef]
- Kaleta, J.; Bastien, G.; Wen, J.; Dračínský, M.; Tortorici, E.; Císařová, I.; Beale, P.D.; Rogers, C.T.; Michl, J. Bulk inclusions of double pyridazine molecular rotors in hexagonal tris(o-phenylene)cyclotriphosphazene. J. Org. Chem. 2019, 84, 8449–8467. [Google Scholar] [CrossRef]
- Sozzani, P.; Bracco, S.; Comotti, A.; Ferretti, L.; Simonutti, R. Methane and carbon dioxide storage in a porous van der Waals crystal. Angew. Chem. Int. Ed. 2005, 44, 1816–1820. [Google Scholar] [CrossRef]
- Allcock, H.R.; Siegel, L.A. Phosphonitrilic compounds. III. Molecular inclusion compounds of tris(o-phenylenedioxy)phosphonitrile trimer. J. Am. Chem. Soc. 1964, 86, 5140–5144. [Google Scholar] [CrossRef]
- Msayib, K.J.; Book, D.; Budd, P.M.; Chaukura, N.; Harris, K.D.M.; Helliwell, M.; Tedds, S.; Walton, A.; Warren, J.E.; Xu, M.C.; et al. Nitrogen and hydrogen adsorption by an organic microporous crystal. Angew. Chem. Int. Ed. 2009, 48, 3273–3277. [Google Scholar] [CrossRef]
- Wuest, J.D. Engineering crystals by the strategy of molecular tectonics. Chem. Commun. 2005, 5830–5837. [Google Scholar] [CrossRef]
- Hosseini, M.W. Molecular tectonics: From simple tectons to complex molecular networks. Acc. Chem. Res. 2005, 38, 313–323. [Google Scholar] [CrossRef]
- Hosseini, M.W. Reflexion on molecular tectonics. CrystEngComm 2004, 6, 318–322. [Google Scholar] [CrossRef]
- Su, D.; Wang, X.; Simard, M.; Wuest, J.D. Molecular tectonics. Supramol. Chem. 1995, 6, 171–178. [Google Scholar] [CrossRef]
- Lin, R.-B.; He, Y.; Li, P.; Wang, H.; Zhou, W.; Chen, B. Multifunctional porous hydrogen-bonded organic framework materials. Chem. Soc. Rev. 2019, 48, 1362–1389. [Google Scholar] [CrossRef] [PubMed]
- Hisaki, I.; Xin, C.; Takahashi, K.; Nakamura, T. Designing hydrogen-bonded organic frameworks (HOFs) with permanent porosity. Angew. Chem. Int. Ed. 2019, 58, 11160–11170. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, J.-W.; Zhang, J.-H.; Lai, S.; Zhong, D.-C. Hydrogen-bonded organic frameworks: Design, structures and potential applications. CrystEngComm 2018, 20, 5884–5898. [Google Scholar] [CrossRef]
- Han, Y.-F.; Yuan, Y.-X.; Wang, H.-B. Porous hydrogen-bonded organic frameworks. Molecules 2017, 22, 266. [Google Scholar] [CrossRef] [Green Version]
- Mastalerz, M.; Oppel, I.M. Rational construction of an extrinsic porous molecular crystal with an extraordinary high specific surface area. Angew. Chem. Int. Ed. 2012, 51, 5252–5255. [Google Scholar] [CrossRef]
- Fournier, J.H.; Maris, T.; Wuest, J.D. Molecular tectonics. Porous hydrogen-bonded networks built from derivatives of 9,9’-spirobifluorene. J. Org. Chem. 2004, 69, 1762–1775. [Google Scholar] [CrossRef]
- Demers, E.; Maris, T.; Wuest, J.D. Molecular tectonics. Porous hydrogen-bonded networks built from derivatives of 2,2’,7,7’-tetraphenyl-9,9’-spirobi[9 H-fluorene]. Cryst. Growth Des. 2005, 5, 1227–1235. [Google Scholar] [CrossRef]
- Chen, T.H.; Popov, I.; Kaveevivitchai, W.; Chuang, Y.C.; Chen, Y.S.; Daugulis, O.; Jacobson, A.J.; Miljanic, O.S. Thermally robust and porous noncovalent organic framework with high affinity for fluorocarbons and CFCs. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Chovnik, O.; Shimon, L.J.W.; Lahav, M.; van der Boom, M.E. Directed molecular structure variations of three-dimensional halogen-bonded organic frameworks (XBOFs). Cryst. Growth Des. 2018, 18, 1967–1977. [Google Scholar] [CrossRef]
- Nikolayenko, V.I.; Castell, D.C.; van Heerden, D.P.; Barbour, L.J. Guest-induced structural transformations in a porous halogen-bonded framework. Angew. Chem. Int. Ed. 2018, 57, 12086–12091. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.T.; Ellington, T.L.; Allen, K.E.; Gorden, J.D.; Rheingold, A.L.; Tschumper, G.S.; Hammer, N.I.; Watkins, D.L. Systematic experimental and computational studies of substitution and hybridization effects in solid-state halogen bonded assemblies. Cryst. Growth Des. 2018, 18, 3244–3254. [Google Scholar] [CrossRef]
- González, L.; Gimeno, N.; Tejedor, R.M.; Polo, V.; Ros, M.B.; Uriel, S.; Serrano, J.L. Halogen-bonding complexes based on bis(iodoethynyl)benzene units: A new versatile route to supramolecular materials. Chem. Mater. 2013, 25, 4503–4510. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Supramolecular hierarchy among halogen-bond donors. Chem. Eur. J. 2013, 19, 16240–16247. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of halogen bonding in solution: Substituent, structural, and solvent effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef]
- Baldrighi, M.; Bartesaghi, D.; Cavallo, G.; Chierotti, M.R.; Gobetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Polymorphs and co-crystals of haloprogin: An antifungal agent. CrystEngComm. 2014, 16, 5897–5904. [Google Scholar] [CrossRef] [Green Version]
- Lemouchi, C.; Vogelsberg, C.S.; Zorina, L.; Simonov, S.; Batail, P.; Brown, S.; Garcia-Garibay, M.A. Ultra-fast rotors for molecular machines and functional materials via halogen bonding: Crystals of 1,4-bis(iodoethynyl)bicyclo[2.2.2]octane with distinct gigahertz rotation at two sites. J. Am. Chem. Soc. 2011, 133, 6371–6379. [Google Scholar] [CrossRef] [Green Version]
- Dunitz, J.D.; Gehrer, H.; Britton, D. The crystal structure of diiodacetylene: An example of pseudosymmetry. Acta Crystallogr. B 1972, 28, 1989–1994. [Google Scholar] [CrossRef]
- Guo, W.Z.; Galoppini, E.; Gilardi, R.; Rydja, G.I.; Chen, Y.H. Weak intermolecular interactions in the crystal structures of molecules with tetrahedral symmetry: Diamondoid nets and other motifs. Cryst. Growth Des. 2001, 1, 231–237. [Google Scholar] [CrossRef]
- Kaleta, J.; Bastien, G.; Císařová, I.; Batail, P.; Michl, J. Molecular rods: Facile desymmetrization of 1,4-diethynylbicyclo[2.2.2]octane. Eur. J. Org. Chem. 2018, 2018, 5137–5142. [Google Scholar] [CrossRef]
- Galoppini, E.; Gilardi, R. Weak hydrogen bonding between acetylenic groups: The formation of diamondoid nets in the crystal structure of tetrakis(4-ethynylphenyl)methane. Chem. Commun. 1999, 173–174. [Google Scholar] [CrossRef]
- Dikundwar, A.G.; Sathishkumar, R.; Guru Row, T.N.; Desiraju, G.R. Structural variability in the monofluoroethynylbenzenes mediated through interactions involving “organic” fluorine. Cryst. Growth Des. 2011, 11, 3954–3963. [Google Scholar] [CrossRef]
- Thakur, T.S.; Sathishkumar, R.; Dikundwar, A.G.; Guru Row, T.N.; Desiraju, G.R. Third polymorph of phenylacetylene. Cryst. Growth Des. 2010, 10, 4246–4249. [Google Scholar] [CrossRef]
- Dziubek, K.; Podsiadło, M.; Katrusiak, A. Nearly isostructural polymorphs of ethynylbenzene: Resolution of ≡CH···π(arene) and cooperative ≡CH···π(C≡C) interactions by pressure freezing. J. Am. Chem. Soc. 2007, 129, 12620–12621. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.K.; Nangia, A. Ethynyl group as a supramolecular halogen and C≡C–H···C≡C trimer synthon in 2,4,6-tris(4-ethynylphenoxy)-1,3,5-triazine. Cryst. Growth Des. 2007, 7, 393–401. [Google Scholar] [CrossRef]
- Ohkita, M.; Suzuki, T.; Nakatani, K.; Tsuji, T. Polar assembly of 2,6-diethynylpyridine through C(sp2)–H···N, C(sp)–H···π and π–π stacking interactions: Crystal structure and nonlinear optical properties. Chem. Lett. 2001, 30, 988–989. [Google Scholar] [CrossRef]
- Robinson, J.M.; Kariuki, B.M.; Harris, K.D.; Philp, D. Interchangeability of halogen and ethynyl substituents in the solid state structures of di- and tri-substituted benzenes. J. Chem. Soc. Perkin Trans. 1998, 2, 2459–2470. [Google Scholar] [CrossRef]
- Weiss, H.-C.; Bläser, D.; Boese, R.; Doughan, B.; Haley, M. C–H···π interactions in ethynylbenzenes: The crystal structures of ethynylbenzene and 1,3,5-triethynylbenzene, and a redetermination of the structure of 1,4-diethynylbenzene. Chem. Commun. 1997, 1703–1704. [Google Scholar] [CrossRef]
- Steiner, T.; Starikov, E.B.; Amado, A.M.; Teixeira-Dias, J.J.C. Weak hydrogen bonding. Part 2. The hydrogen bonding nature of short C–H···π contacts: Crystallographic, spectroscopic and quantum mechanical studies of some terminal alkynes. J. Chem. Soc. Perkin Trans. 1995, 2, 1321–1326. [Google Scholar] [CrossRef]
- Barbour, L.J. Crystal porosity and the burden of proof. Chem. Commun. 2006, 11, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, C.A.; Đaković, M.; Aakeröy, C.B. Diamondoid architectures from halogen-bonded halides. Chem. Commun. 2018, 54, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Wavefunction, Inc. Irvine, CA 92612, USA. Available online: https://www.wavefun.com/spartan (accessed on 8 March 2020).
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | C–I⋯O | d(I⋯O)/Å | ND a | %RR b | ∠(C–I⋯O)/° |
|---|---|---|---|---|---|
| I4TEPM | C9–I10···C8(π) c | 3.405(12) | 0.925 | 7.47 | 165.5(4) |
| C9–I10···C9(π) c | 3.266(13) | 0.888 | 11.2 | 173.6(5) | |
| I4TEPM·2THF | C9–I10···O11 d | 2.965(5) | 0.847 | 15.3 | 170.1(3) |
| I4TEPM·2DMSO | C9–I10···O23 e | 3.013(3) | 0.861 | 13.9 | 162.0(14) |
| C18–I19···O23 f | 2.797(3) | 0.799 | 20.1 | 170.0(14) | |
| I4TEPM·2Dioxane | C1–I1···O1 | 2.773(4) | 0.792 | 20.8 | 174.3(11) |
| C17–I2···O2 g | 2.819(3) | 0.805 | 19.5 | 174.4(9) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunawardana, C.A.; Sinha, A.S.; Reinheimer, E.W.; Aakeröy, C.B. From Frustrated Packing to Tecton-Driven Porous Molecular Solids. Chemistry 2020, 2, 179-192. https://doi.org/10.3390/chemistry2010011
Gunawardana CA, Sinha AS, Reinheimer EW, Aakeröy CB. From Frustrated Packing to Tecton-Driven Porous Molecular Solids. Chemistry. 2020; 2(1):179-192. https://doi.org/10.3390/chemistry2010011
Chicago/Turabian StyleGunawardana, Chamara A., Abhijeet S. Sinha, Eric W. Reinheimer, and Christer B. Aakeröy. 2020. "From Frustrated Packing to Tecton-Driven Porous Molecular Solids" Chemistry 2, no. 1: 179-192. https://doi.org/10.3390/chemistry2010011
APA StyleGunawardana, C. A., Sinha, A. S., Reinheimer, E. W., & Aakeröy, C. B. (2020). From Frustrated Packing to Tecton-Driven Porous Molecular Solids. Chemistry, 2(1), 179-192. https://doi.org/10.3390/chemistry2010011