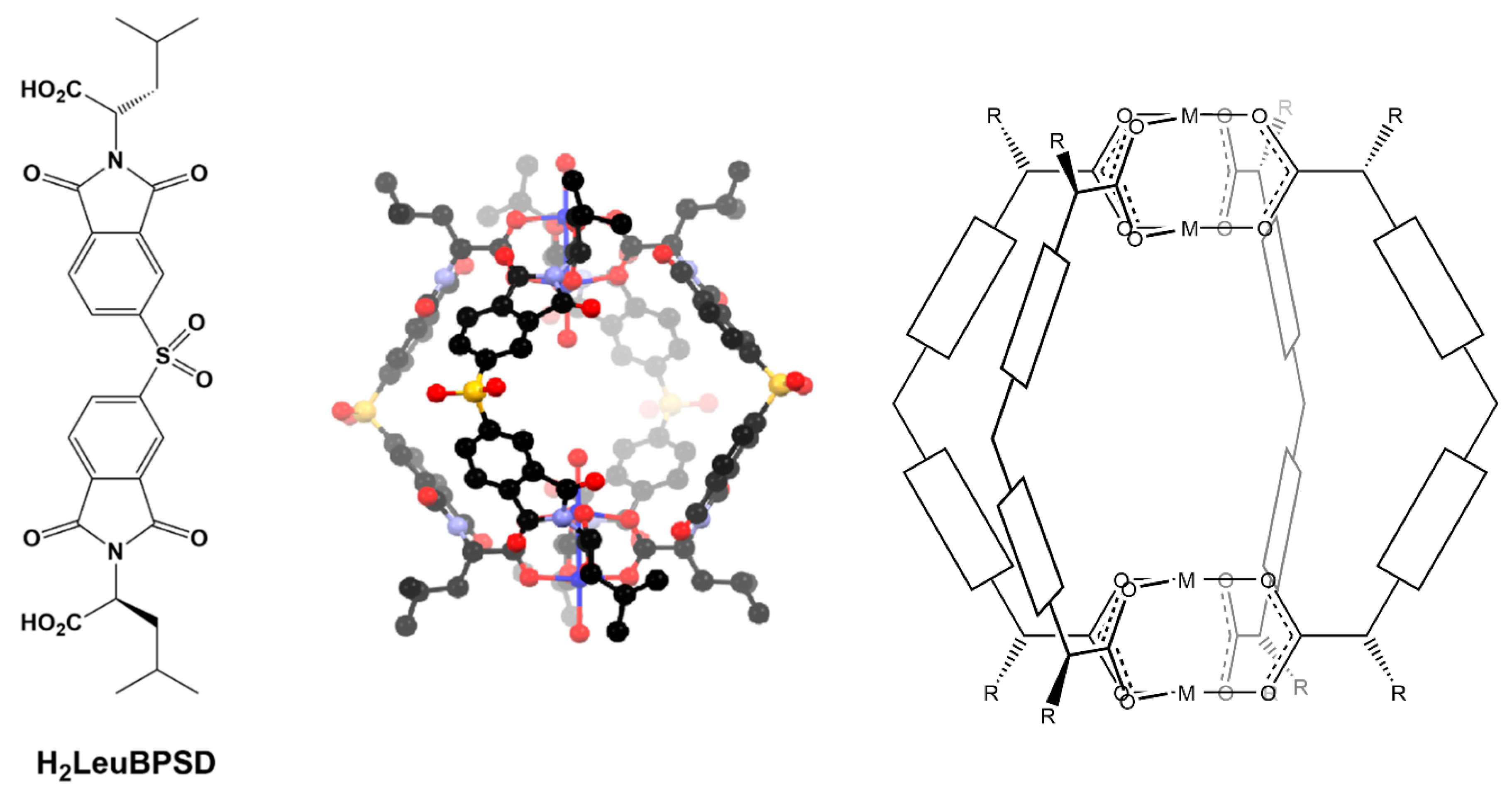
Towards a Generalized Synthetic Strategy for Variable Sized Enantiopure M4L4 Helicates



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Details
2.2. Synthesis and Characterisation
2.3. X-ray Crystallography
3. Results
3.1. Design and Synthesis of Ligands
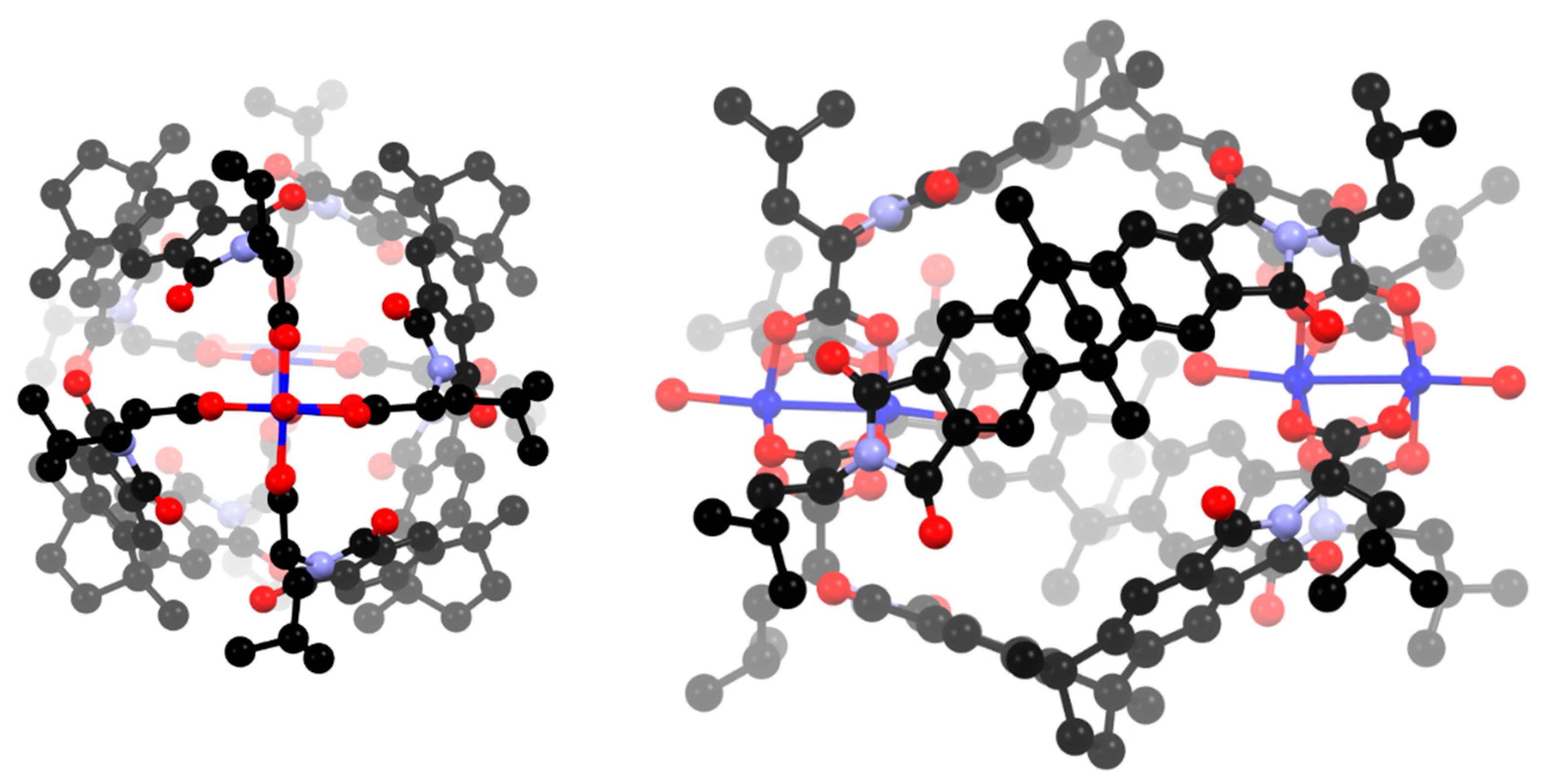
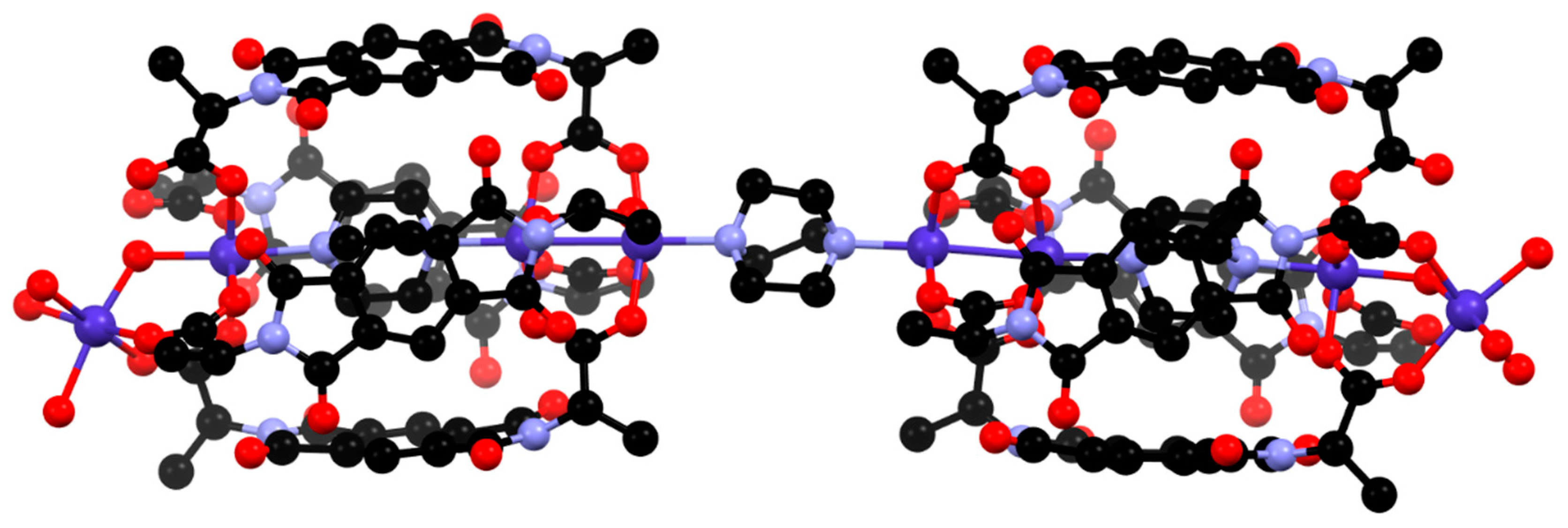
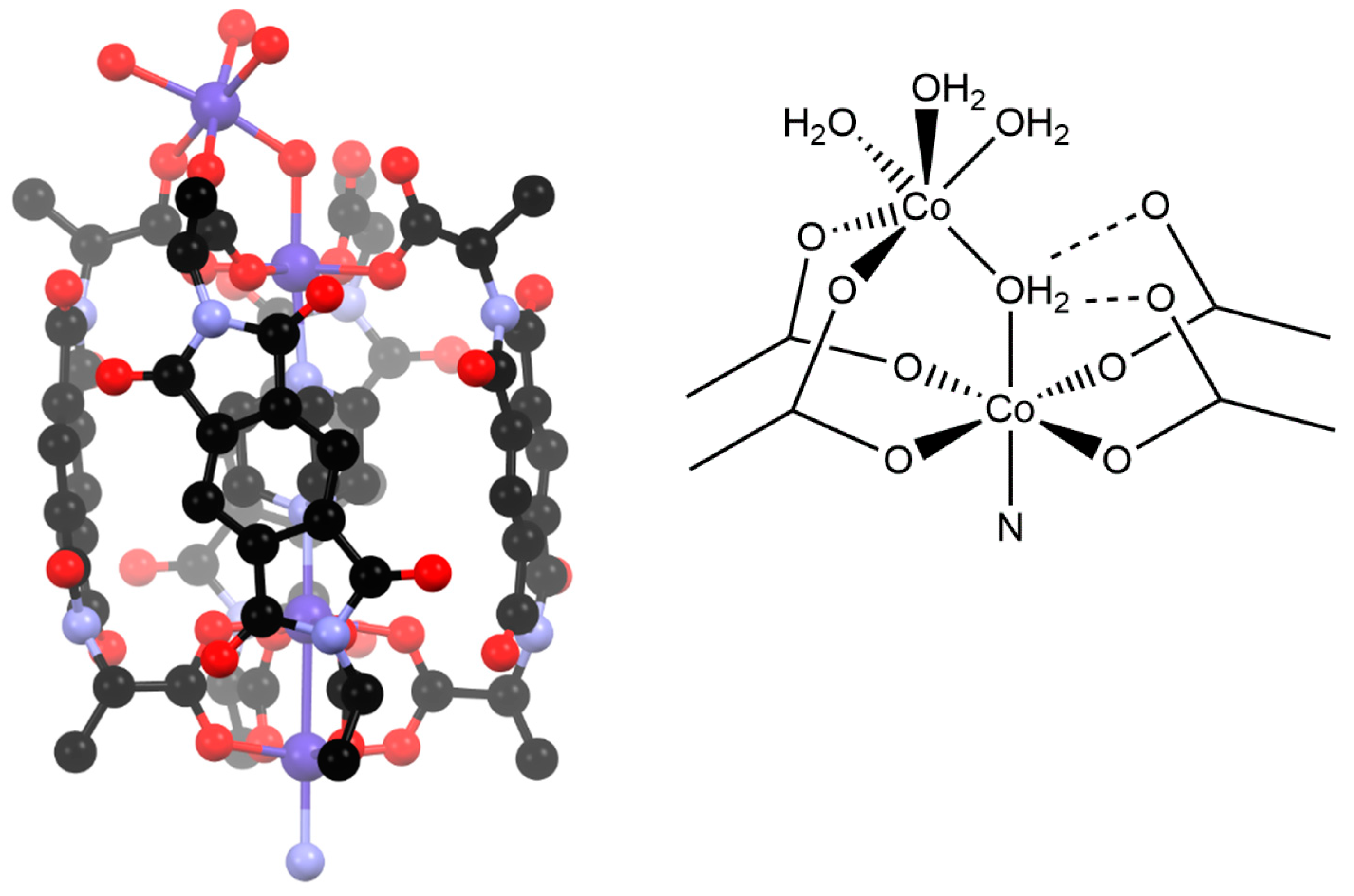
3.2. Synthesis and Structure of Helicate-Type Complexes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Crystallographic Data and Special Refinement Details
References
- Cram, D.J.; Cram, J.M. Container Molecules and Their Guests; Royal Society of Chemistry: Cambridge, UK, 1997. [Google Scholar]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011, 111, 6810–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarra, S.; Wood, D.M.; Roberts, D.A.; Nitschke, J.R. Molecular containers in complex chemical systems. Chem. Soc. Rev. 2015, 44, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friese, V.A.; Kurth, D.G. From coordination complexes to coordination polymers through self-assembly. Curr. Opinon Colloid Interface Sci. 2009, 14, 81–93. [Google Scholar] [CrossRef]
- Vardhan, H.; Yusubov, M.; Verpoort, F. Self-assembled metal-organic polyhedra: An overview of various applications. Coord. Chem. Rev. 2016, 306, 171–194. [Google Scholar] [CrossRef]
- Saha, M.L.; De, S.; Pramanik, S.; Schmittel, M. Orthogonality in discrete self-assembly—Survey of current concepts. Chem. Soc. Rev. 2013, 42, 6860–6909. [Google Scholar] [CrossRef]
- Northrop, B.H.; Zheng, Y.-R.; Chi, K.-W.; Stang, P.J. Self-Organization in Coordination-Driven Self-Assembly. Acc. Chem. Res. 2009, 42, 1554–1563. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Woods, B.; Wenzel, M. The Promise of Self-Assembled 3D Supramolecular Coordination Complexes for Biomedical Applications. Inorg. Chem. 2017, 56, 14715–14729. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-J.; Yang, H.-B.; Shionoya, M. Chiral metallosupramolecular architectures. Chem. Soc. Rev. 2017, 46, 2555–2576. [Google Scholar] [CrossRef]
- Clegg, J.K.; McMurtrie, J.C. Chiral Metallosupramolecular Polyhedra. In Chirality in Supramolecular Assemblies: Causes and Consequences; Keene, F.R., Ed.; John Wiley & Sons: Chichester, UK, 2016; pp. 218–256. [Google Scholar]
- Pan, M.; Wu, K.; Zhang, J.-H.; Su, C.-Y. Chiral metal–organic cages/containers (MOCs): From structural and stereochemical design to application. Coord. Chem. Rev. 2019, 378, 333–349. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Xie, S.-M.; Zi, M.; Yuan, L.-M. Recent advances of application of porous molecular cages for enantioselective recognition and separation. J. Sep. Sci. 2020, 43, 134–149. [Google Scholar] [CrossRef]
- Saalfrank, R.W.; Glaser, H.; Demleitner, B.; Hampel, F.; Chowdhry, M.M.; Schünemann, V.; Trautwein, A.X.; Vaughan, G.B.M.; Yeh, R.; Davis, A.V.; et al. Self-Assembly of Tetrahedral and Trigonal Antiprismatic Clusters [Fe4(L4)4] and [Fe6(L5)6] on the Basis of Trigonal Tris-Bidentate Chelators. Chem. Eur. J. 2002, 8, 493–497. [Google Scholar] [CrossRef]
- Zhao, C.; Toste, F.D.; Raymond, K.N.; Bergman, R.G. Nucleophilic Substitution Catalyzed by a Supramolecular Cavity Proceeds with Retention of Absolute Stereochemistry. J. Am. Chem. Soc. 2014, 136, 14409–14412. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-P.; Yuan, L.-B.; Song, J.-S.; Chen, G.-H.; Lin, Q.; Li, C.; Zhang, L.; Zhang, J. Optical Resolution of the Water-Soluble Ti4(embonate)6 Cages for Enantioselective Recognition of Chiral Drugs. Chem. Mater. 2018, 30, 7769–7775. [Google Scholar] [CrossRef]
- Browne, C.; Brenet, S.; Clegg, J.K.; Nitschke, J.R. Solvent-Dependent Host–Guest Chemistry of an Fe8L12 Cubic Capsule. Angew. Chem. Int. Ed. 2013, 52, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Y.; Jia, J.-H.; Zheng, X.-Y.; Zhang, Q.; Xiong, K.-C.; Zhang, Z.-M.; Wang, Q.-M. Enantiopure Magnetic Heterometallic Coordination Cubic Cages [MII8CuII6] (M = Ni, Co). Cryst. Growth Des. 2018, 18, 4555–4561. [Google Scholar] [CrossRef]
- Guo, J.; Xu, Y.-W.; Li, K.; Xiao, L.-M.; Chen, S.; Wu, K.; Chen, X.-D.; Fan, Y.-Z.; Liu, J.-M.; Su, C.-Y. Regio- and Enantioselective Photodimerization within the Confined Space of a Homochiral Ruthenium/Palladium Heterometallic Coordination Cage. Angew. Chem. Int. Ed. 2017, 56, 3852–3856. [Google Scholar] [CrossRef]
- Wang, X.; Peng, P.; Xuan, W.; Wang, Y.; Zhuang, Y.; Tian, Z.; Cao, X. Narcissistic chiral self-sorting of molecular face-rotating polyhedral. Org. Biomol. Chem. 2018, 16, 34–37. [Google Scholar] [CrossRef]
- Schweiger, M.; Seidel, S.R.; Schmitz, M.; Stang, P.J. Rational Design of Chiral Nanoscale Adamantanoids. Org. Lett. 2000, 2, 1255–1257. [Google Scholar] [CrossRef]
- Dong, J.; Zhou, Y.; Zhang, F.; Cui, Y. A Highly Fluorescent Metallosalen-Based Chiral Cage for Enantioselective Recognition and Sensing. Chem. Eur. J. 2014, 20, 6455–6461. [Google Scholar] [CrossRef]
- Sun, B.; Nurttila, S.S.; Reek, J.N.H. Synthesis and Characterization of Self-Assembled Chiral FeII2L3 Cages. Chem. Eur. J. 2018, 24, 14693–14700. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Jiao, J.; Li, Z.; Liu, Y.; Han, X.; Cui, Y. Design and Assembly of a Chiral Metallosalen-Based Octahedral Coordination Cage for Supramolecular Asymmetric Catalysis. Angew. Chem. Int. Ed. 2018, 57, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, X.; Xuan, W.; Peng, P.; Li, Z.; Lu, R.; Wu, S.; Tian, Z.; Cao, X. Chiral separation and characterization of triazatruxene-based face-rotating polyhedral: The role of non-covalent facial interactions. Chem. Commun. 2018, 54, 4685–4688. [Google Scholar] [CrossRef] [PubMed]
- Bhat, I.A.; Devaraj, A.; Howlader, P.; Chi, K.-W.; Mukherjee, P.S. Preparation of a chiral Pt12 tetrahedral cage and its use in catalytic Michael addition reaction. Chem. Commun. 2018, 54, 4814–4817. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Peschard, S.; Horiuchi, S.; Nishioka, Y.; Fujita, M. Remote chiral transfer into [2 + 2] and [2 + 4] cycloadditions within self-assembled molecular flasks. Supramol. Chem. 2011, 23, 199–208. [Google Scholar] [CrossRef]
- Argent, S.P.; Riis-Johannessen, T.; Jeffery, J.C.; Harding, L.P.; Ward, M.D. Diastereoselective formation and optical activity of an M4L6 cage complex. Chem. Commun. 2005, 4647–4649. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Burk, S.; Weis, P. Chiral Confined Space: Induction of Stereochemistry in a M4L4 Metallosupramolecular Container. Synthesis 2008, 2963–2967. [Google Scholar] [CrossRef]
- Ren, D.-H.; Qiu, D.; Pang, C.-Y.; Li, Z.; Gu, Z.-G. Chiral tetrahedral iron(II) cages: Diastereoselective subcomponent self-assembly, structure interconversion and spin-crossover properties. Chem. Commun. 2015, 51, 788–791. [Google Scholar] [CrossRef]
- Ousaka, N.; Clegg, J.K.; Nitschke, J.R. Nonlinear Enhancement of Chiroptical Response through Subcomponent Substitution in M4L6 Cages. Angew. Chem. Int. Ed. 2012, 51, 1464–1468. [Google Scholar] [CrossRef]
- Yan, L.-L.; Tan, C.-H.; Zhang, G.-L.; Zhou, L.-P.; Bünzli, J.-C.; Sun, Q.-F. Stereocontrolled Self-Assembly and Self-Sorting of Luminescent Europium Tetrahedral Cages. J. Am. Chem. Soc. 2015, 137, 8550–8555. [Google Scholar] [CrossRef]
- Wisser, B.; Chamayou, A.-C.; Miller, R.; Scherer, W.; Janiak, C. A chiral C3-symmetric hexanuclear triangular-prismatic copper(ii) cluster derived from a highly modular dipeptidic N,N′-terephthaloyl-bis(S-aminocarboxylato) ligand. CrystEngComm 2008, 10, 461–464. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Wu, A.; Liang, Y.; Wang, X.; Liang, F. Synthesis, structure and properties of an octahedral dinuclear-based Cu12 nanocage of trimesoyltri(l-alanine). RSC Adv. 2016, 6, 9911–9915. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Shi, W.; Niu, Z.; Li, H.-H.; Zhao, B.; Cheng, P.; Liao, D.-Z.; Yan, S.-P. A new type of polyhedron-based metal–organic frameworks with interpenetrating cationic and anionic nets demonstrating ion exchange, adsorption and luminescent properties. Chem. Commun. 2011, 47, 6425–6427. [Google Scholar] [CrossRef]
- Boer, S.A.; Turner, D.R. Self-selecting homochiral quadruple-stranded helicates and control of supramolecular chirality. Chem. Commun. 2015, 51, 17375–17378. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Kang, J.; Cui, H.; Wang, Y.; Liu, L.; Zhang, L.; Su, C.-Y. Homochiral coordination cages assembled from dinuclear paddlewheel nodes and enantiopure ditopic ligands: Syntheses, structures and catalysis. Dalton Trans. 2015, 44, 12180–12188. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yang, H.; Wang, Z.U.; Liu, Y.; Zhou, H.-C.; Li, J.-R. Unusual preservation of polyhedral molecular building units in a metal–organic framework with evident desymmetrization in ligand design. Chem. Commun. 2014, 50, 563–565. [Google Scholar] [CrossRef]
- Lu, W.; Yuan, D.; Yakovenko, A.; Zhou, H.-C. Surface functionalization of metal–organic polyhedron for homogeneous cyclopropanation catalysis. Chem. Commun. 2011, 47, 4968–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-R.; Yu, J.; Lu, W.; Sun, L.-B.; Sculley, J.; Balbuena, P.B.; Zhou, H.-C. Porous materials with pre-designed single-molecule traps for CO2 selective adsorption. Nat. Commun. 2013, 4, 1538. [Google Scholar] [CrossRef]
- Prakash, M.J.; Oh, M.; Liu, X.; Han, K.N.; Seong, G.H.; Lah, M.S. Edge-directed [(M2)2L4] tetragonal metal–organic polyhedra decorated using a square paddle-wheel secondary building unit. Chem. Commun. 2010, 46, 2049–2051. [Google Scholar] [CrossRef]
- Craig, G.A.; Larpent, P.; Kusaka, S.; Matsuda, R.; Kitagawa, S.; Furukawa, S. Switchable gate-opening effect in metal–organic polyhedra assemblies through solution processing. Chem. Sci. 2018, 9, 6463–6469. [Google Scholar] [CrossRef] [Green Version]
- Brega, V.; Zeller, M.; He, Y.; Lu, H.P.; Klosterman, J.K. Multi-responsive metal–organic lantern cages in solution. Chem. Commun. 2015, 51, 5077–5080. [Google Scholar] [CrossRef]
- Boer, S.A.; White, K.F.; Slater, B.; Emerson, A.J.; Knowles, G.P.; Donald, W.A.; Thornton, A.W.; Ladewig, B.P.; Bell, T.D.M.; Hill, M.R.; et al. A Multifunctional, Charge-Neutral, Chiral Octahedral M12L12 Cage. Chem. Eur. J. 2019, 25, 8489–8493. [Google Scholar] [CrossRef]
- Mollick, S.; Mukherjee, S.; Kim, D.; Zhiwei, Q.; Desai, A.V.; Saha, R.; More, Y.D.; Jiang, J.; Lah, M.S.; Ghosh, S.K. Hydrophobic Shielding of Outer Surface: Enhancing the Chemical Stability of Metal–Organic Polyhedra. Angew. Chem. Int. Ed. 2019, 58, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Boer, S.A.; Turner, D.R. Metallosupramolecular Architectures of Ambivergent Bis(Amino Acid) Biphenyldiimides. Chem. Asian J. 2019, 14, 2853–2860. [Google Scholar] [CrossRef]
- Boer, S.A.; Cox, R.P.; Beards, M.J.; Wang, H.; Donald, W.A.; Bell, T.D.M.; Turner, D.R. Elucidation of naphthalene diimide metallomacrocycles and catenanes by solvent dependent excimer and exciplex emission. Chem. Commun. 2019, 55, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Kyratzis, N.; Cao, W.; Izogordina, E.I.; Turner, D.R. Structural changes in coordination polymers in response to small changes in steric bulk (H vs. Me): An experimental and theoretical study. CrystEngComm 2018, 20, 4115–4126. [Google Scholar] [CrossRef]
- Boer, S.A.; Turner, D.R. A robust metallomacrocyclic motif for the formation of interpenetrated coordination polymers. CrystEngComm 2017, 19, 2402–2412. [Google Scholar] [CrossRef]
- Boer, S.A.; Turner, D.R. Interpenetration in π-rich mixed-ligand coordination polymers. Cryst. Growth Des. 2016, 16, 6294–6303. [Google Scholar] [CrossRef]
- Boer, S.A.; Hawes, C.S.; Turner, D.R. Engineering entanglement: Controlling the formation of polycatenanes and polyrotaxanes using π interactions. Chem. Commun. 2014, 50, 1125–1127. [Google Scholar] [CrossRef]
- Rogan, Y.; Malpass-Evans, R.; Carta, M.; Lee, M.; Jansen, J.C.; Bernardo, P.; Clarizia, G.; Tocci, E.; Friess, K.; Lanc, M.; et al. A highly permeable polyimide with enhanced selectivity for membrane gas separations. J. Mater. Chem. A 2014, 2, 4874–4877. [Google Scholar] [CrossRef] [Green Version]
- Faghihi, K.; Moghanian, H. ynthesis and Characterization of New Optically Active Poly(amide-imide)s Based on N,N′-(Pyromellitoyl)-bis-L-Amino Acids and 1,3,4-Oxadiazole Moieties. Des. Monomers Polym. 2010, 13, 207–220. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Rad. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.M.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Rad. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Cryst. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Prasad, T.K.; Hong, D.H.; Suh, M.P. High Gas Sorption and Metal-Ion Exchange of Microporous Metal–Organic Frameworks with Incorporated Imide Groups. Chem. Eur. J. 2010, 16, 14043–14050. [Google Scholar] [CrossRef]
- Leong, C.F.; Faust, T.B.; Turner, P.; Usov, P.M.; Kepert, C.J.; Babarao, R.; Thornton, A.W.; D’Alessandro, D.M. Enhancing selective CO2 adsorption via chemical reduction of a redox-active metal–organic framework. Dalton Trans. 2013, 42, 9831–9839. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-B.; Yang, Q.-Y.; Pan, R.-K.; Liu, S.-G. Diverse cobalt(ii) coordination polymers for water/ethanol separation and luminescence for water sensing applications. CrystEngComm 2018, 20, 3891–3897. [Google Scholar] [CrossRef]
- Li, G.-B.; Liu, J.-M.; Cai, Y.-P.; Su, C.-Y. Structural Diversity of a Series of Mn(II), Cd(II), and Co(II) Complexes with Pyridine Donor Diimide Ligands. Cryst. Growth Des. 2011, 11, 2763–2772. [Google Scholar] [CrossRef]
- Kang, G.; Jeon, Y.; Lee, K.Y.; Kim, J.; Kim, T.H. Reversible Luminescence Vapochromism and Crystal-to-Amorphous-to-Crystal Transformations of Pseudopolymorphic Cu(I) Coordination Polymers. Cryst. Growth Des. 2015, 15, 5183–5187. [Google Scholar] [CrossRef]
- Nakagaki, T.; Harano, A.; Fuchigami, Y.; Tanaka, E.; Kidoaki, S.; Okuda, T.; Iwanaga, T.; Goto, K.; Shinmyozu, T. Formation of Nanoporous Fibers by the Self-Assembly of a Pyromellitic Diimide-Based Macrocycle. Angew. Chem. Int. Ed. 2010, 49, 9676–9679. [Google Scholar] [CrossRef]
- Nalluri, S.K.M.; Zhou, J.; Cheng, T.; Liu, Z.; Nguyen, M.T.; Chen, T.; Patel, H.A.; Krzyaniak, M.D.; Goddard, W.A.; Wasielewski, M.R.; et al. Discrete Dimers of Redox-Active and Fluorescent Perylene Diimide-Based Rigid Isosceles Triangles in the Solid State. J. Am. Chem. Soc. 2018, 141, 1290–1303. [Google Scholar] [CrossRef] [Green Version]
- Colquhoun, H.M.; Zhu, Z.; Williams, D.J.; Drew, M.G.B.; Cardin, C.J.; Gan, Y.; Crawford, A.G.; Marder, T.B. Induced-Fit Binding of π-Electron-Donor Substrates to Macrocyclic Aromatic Ether Imide Sulfones: A Versatile Approach to Molecular Assembly. Chem. Eur. J. 2010, 16, 907–918. [Google Scholar] [CrossRef]
- Liu, Z.-M.; Liu, Y.; Zheng, S.-R.; Yu, Z.-Q.; Pan, M.; Su, C.-Y. Assembly of Trigonal and Tetragonal Prismatic Cages from Octahedral Metal Ions and a Flexible Molecular Clip. Inorg. Chem. 2007, 46, 5814–5816. [Google Scholar] [CrossRef]
- Zhang, Q.; Hamilton, D.G.; Feeder, N.; Teat, S.J.; Goodman, J.M.; Sanders, J.K.M. Synthesis and post-assembly modification of some functionalised, neutral π-associated [2]catenanes. New J. Chem. 1999, 23, 897–903. [Google Scholar] [CrossRef]
- Iwanaga, T.; Nakamoto, R.; Yasutake, M.; Takemura, H.; Sako, K.; Shinmyozu, T. Cyclophanes within Cyclophanes: The Synthesis of a Pyromellitic Diimide-Based Macrocycle as a Structural Unit in a Molecular Tube and Its Inclusion Phenomena. Angew. Chem. Int. Ed. 2006, 45, 3643–3647. [Google Scholar] [CrossRef]
- Ge, C.; Li, X.; Zhang, X.; Zhao, Y.; Zhang, R. 2,2′-(1,3,5,7-Tetra oxo-1,2,3,5,6,7-hexa-hydro-pyrrolo[3,4-f]isoindole-2,6-di-yl)diacetic acid N,N-dimethyl formamide disolvate. Acta. Crystallogr. Sect. E 2009, 65, o2400. [Google Scholar] [CrossRef] [Green Version]
- Barooah, N.; Sarma, R.J.; Baruah, J.B. Solid-state hydrogen bonded assembly of N,N′-bis(glycinyl)-pyromellitic diimide with aromatic guests. CrystEngComm 2006, 8, 608–615. [Google Scholar] [CrossRef]
- Barooah, N.; Singh, W.M.; Baruah, J.B. Preferential deprotonation and conformational stability of dicarboxylic acids: A packing effect. J. Mol. Struc. 2008, 875, 329–338. [Google Scholar] [CrossRef]
- Ge, C.-H.; Zhang, X.-D.; Zhang, H.-D.; Zhao, Y.; Li, X.-Q.; Zhang, R. Color Variety of Organic Salt of N,N′-Bis(Glycinyl)Pyromellitic Diimide and N-Containing Base. Mol. Cryst. Liq. Cryst. 2011, 534, 114–123. [Google Scholar] [CrossRef]
- Barooah, N.; Baruah, J.B. Effect of phenyl group on the self assembly of N,N′-bis-(2-phenylglycinyl)pyromellitic diimide with aromatic hydrocarbons. J. Mol. Struc. 2008, 872, 205–211. [Google Scholar] [CrossRef]
- Jana, P.; Maity, S.; Maity, S.K.; Ghorai, P.K.; Halder, D. Insights into H-aggregates and CH⋯O hydrogen bond mediated self-assembly of pyromellitic bisimide. CrystEngComm 2012, 14, 6586–6592. [Google Scholar] [CrossRef]
- Barooah, N.; Sarma, R.J.; Batsanov, A.S.; Baruah, J.B. Structural aspects of adducts of N-phthaloylglycine and its derivatives. J. Mol. Struc. 2006, 791, 122–130. [Google Scholar] [CrossRef]
- Joarder, B.; Mukherjee, S.; Chaudhari, A.K.; Desai, A.V.; Manna, B.; Ghosh, S.K. Guest-Responsive Function of a Dynamic Metal–Organic Framework with a π Lewis Acidic Pore Surface. Chem. Eur. J. 2014, 20, 15303–15308. [Google Scholar] [CrossRef]
- Thuéry, P.; Masci, B. Self-assembly of an Octa-uranate Cage Complex with a Rigid bis-Catechol Ligand. Supramol. Chem. 2003, 15, 95–99. [Google Scholar] [CrossRef]
- Abrahams, B.F.; FitzGerald, N.J.; Robson, R. A Doughnut-Like (MnIII)12 Metallocycle Formed by a Rigid Angular Bis-Catecholate with a Nanometer-Sized Central Hole. Inorg. Chem. 2010, 49, 5953–5956. [Google Scholar] [CrossRef]
- Kawakami, Y.; Ogishima, T.; Kawara, T.; Yamauchi, S.; Okamoto, K.; Nikaido, S.; Souma, D.; Jim, R.-H.; Kabe, Y. Silane catecholates: Versatile tools for self-assembled dynamic covalent bond chemistry. Chem. Commun. 2019, 55, 6066–6069. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boer, S.A.; Cao, W.; Glascott, B.K.; Turner, D.R. Towards a Generalized Synthetic Strategy for Variable Sized Enantiopure M4L4 Helicates. Chemistry 2020, 2, 613-625. https://doi.org/10.3390/chemistry2030038
Boer SA, Cao W, Glascott BK, Turner DR. Towards a Generalized Synthetic Strategy for Variable Sized Enantiopure M4L4 Helicates. Chemistry. 2020; 2(3):613-625. https://doi.org/10.3390/chemistry2030038
Chicago/Turabian StyleBoer, Stephanie A., Winnie Cao, Bianca K. Glascott, and David R. Turner. 2020. "Towards a Generalized Synthetic Strategy for Variable Sized Enantiopure M4L4 Helicates" Chemistry 2, no. 3: 613-625. https://doi.org/10.3390/chemistry2030038
APA StyleBoer, S. A., Cao, W., Glascott, B. K., & Turner, D. R. (2020). Towards a Generalized Synthetic Strategy for Variable Sized Enantiopure M4L4 Helicates. Chemistry, 2(3), 613-625. https://doi.org/10.3390/chemistry2030038