
A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide



Abstract
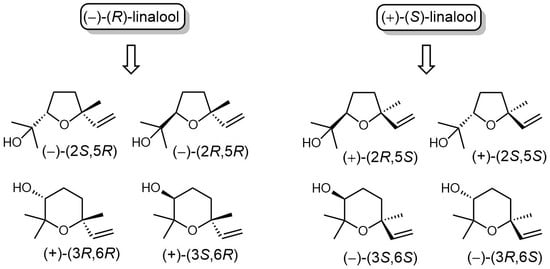
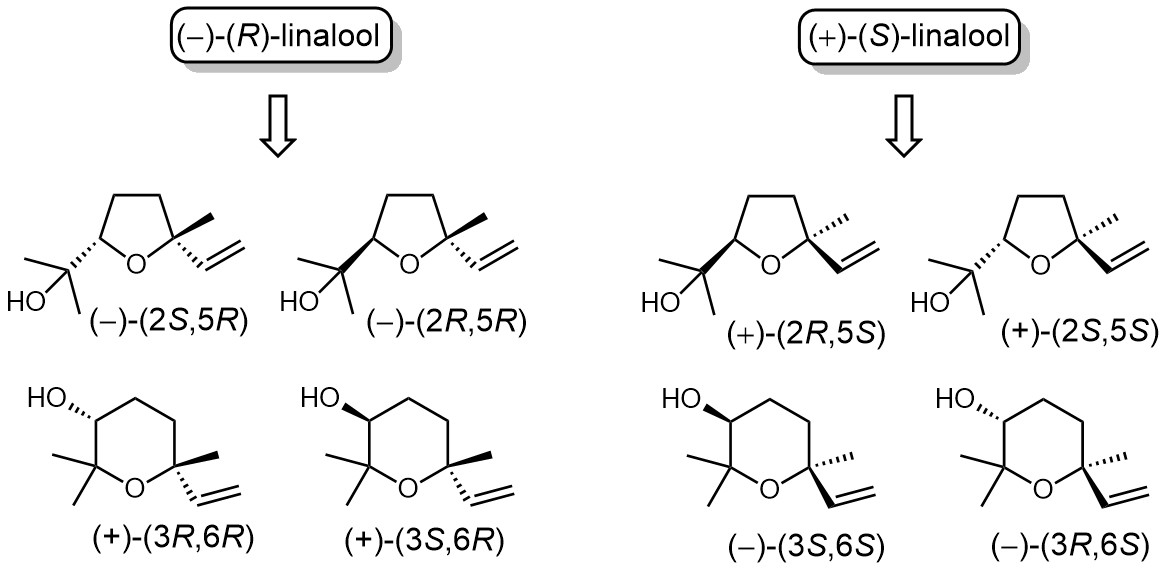
:
1. Introduction
2. Materials and Methods
2.1. Materials and General Methods
2.2. Analytical Methods and Characterization of the Chemical Compounds
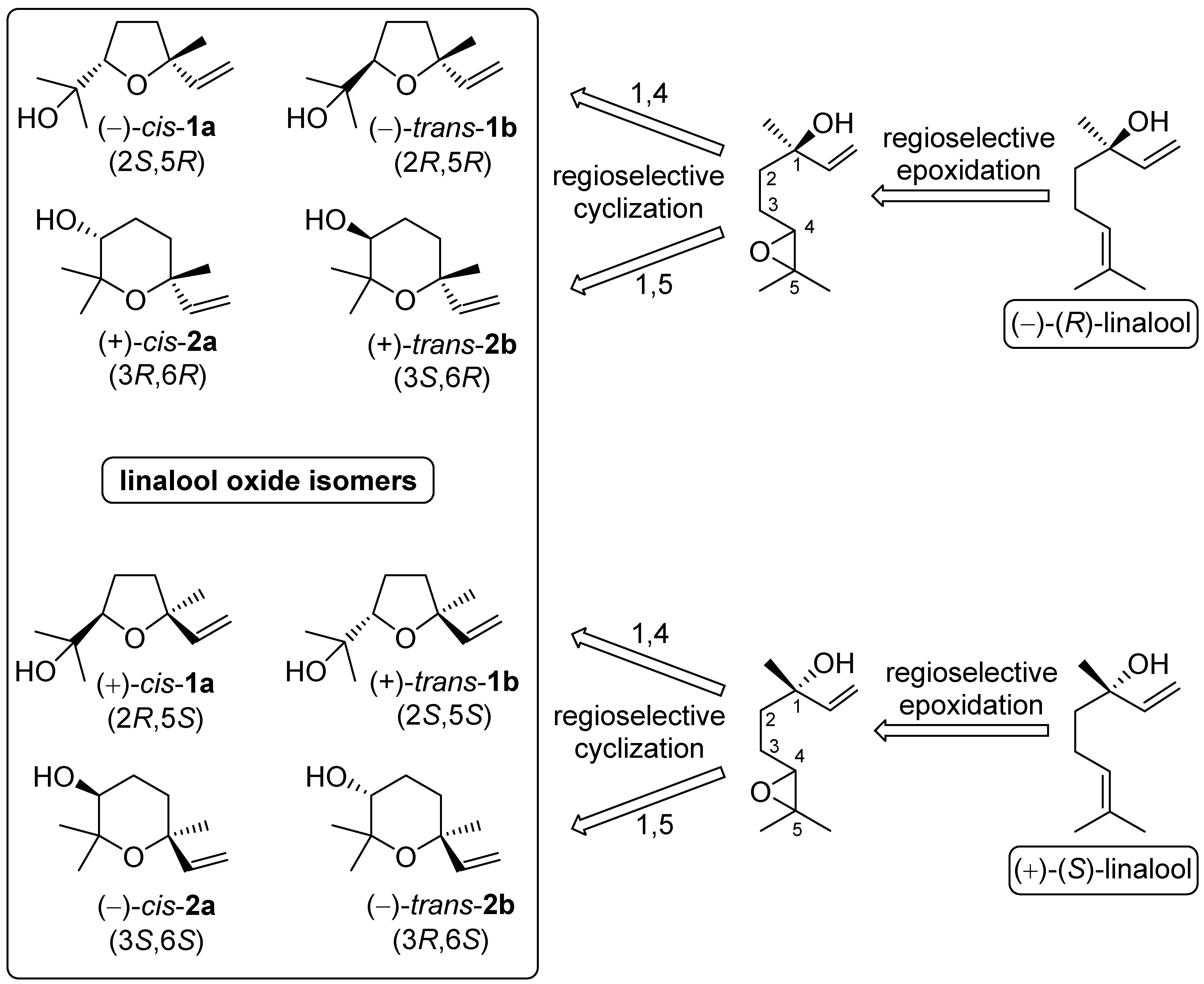
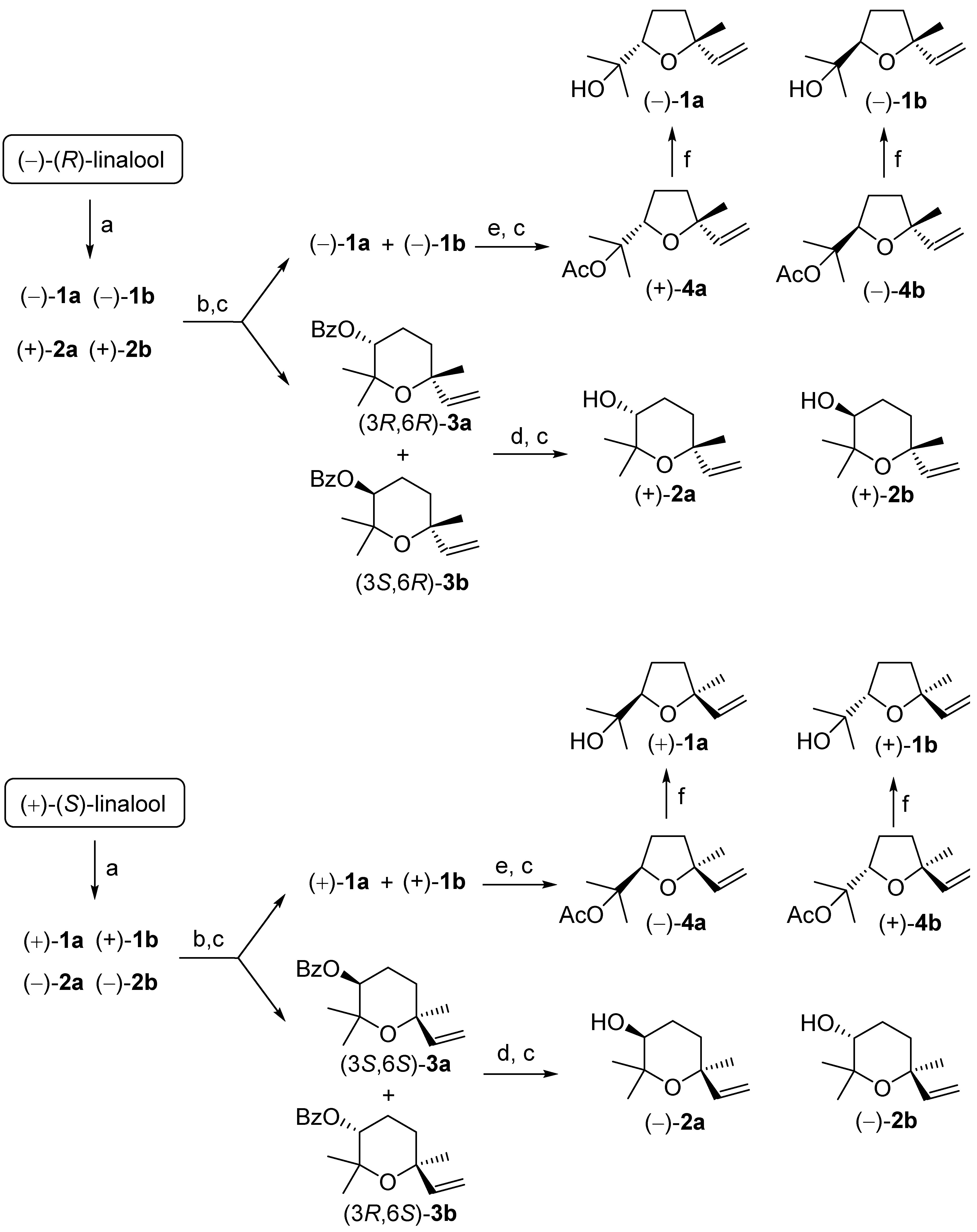
2.3. Procedure for the Preparation of the Linalool Oxide Isomers (2S,5R)-1a, (2R,5R)-1b, (3R,6R)-2a and (3S,6R)-2b
2.3.1. Preparation of the Mixture of the Linalool Oxide Isomers 1a, 1b, 2a and 2b from (R)-Linalool
2.3.2. Procedure for the Separation of the Linalool Oxide Isomers
2.4. Procedure for the Preparation of the Linalool Oxide Isomers (2R,5S)-1a, (2S,5S)-1b, (3S,6S)-2a and (3R,6S)-2b
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragrance and Flavor Materials, 4th ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; p. 145. [Google Scholar]
- Winterhalter, P.; Katzenberger, D.; Schreier, P. 6,7-Epoxy-linalool and related oxygenated terpenoids from Carica papaya fruit. Phytochemistry 1986, 25, 1347–1350. [Google Scholar] [CrossRef]
- Aprotosoaie, A.C.; Hăncianu, M.; Costache, I.-I.; Miron, A. Linalool: A review on a key odorant molecule with valuable biological properties. Flavour Fragr. J. 2014, 29, 193–219. [Google Scholar] [CrossRef]
- Wright, J. Linalool oxide. Perfum. Flavorist 2013, 38, 18–19. [Google Scholar]
- Stevens, K.L.; Bomben, J.; Lee, A.; McFadden, W.H. Volatiles from grapes. Muscat of Alexandria. J. Agric. Food Chem. 1966, 14, 249–252. [Google Scholar] [CrossRef]
- Flath, R.A.; Forrey, R.R. Volatile components of papaya (Carica papaya L., Solo variety). J. Agric. Food Chem. 1977, 25, 103–109. [Google Scholar] [CrossRef]
- Williams, P.J.; Strauss, C.R.; Wilson, B. New linalool derivatives in muscat of Alexandria grapes and wines. Phytochemistry 1980, 19, 1137–1139. [Google Scholar] [CrossRef]
- Wang, D.; Ando, K.; Morita, K.; Kubota, K.; Kobayashi, A. Optical isomers of linalool and linalool oxides in tea aroma. Biosci. Biotech. Biochem. 1994, 58, 2050–2053. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.-H.; Watanabe, N.; Ijima, Y.; Yagi, A.; Sakata, K. Cis- and trans-linalool 3,7-oxides and methyl salicylate glycosides and (Z)-3-hexenyl β-D-glucopyranoside as aroma precursors from tea leaves for oolong tea. Biosci. Biotech. Biochem. 1996, 60, 1815–1819. [Google Scholar] [CrossRef]
- Jiang, L.; Kojima, H.; Yamada, K.; Kobayashi, A.; Kubota, K. Isolation of some glycosides as aroma precursors in young leaves of japanese pepper (Xanthoxylum piperitum DC.). J. Agric. Food Chem. 2001, 49, 5888–5894. [Google Scholar] [CrossRef] [PubMed]
- Chanotiya, C.S.; Sammal, S.S.; Mathela, C.S. Composition of a new chemotype of Tanacetum nubigenum. Indian J. Chem. B 2005, 44, 1922–1926. [Google Scholar] [CrossRef]
- Gajula, S.; Madhu, M.; Chintakrinda, S.K.; Yadav, J.S.; Mohapatra, D.K. A protecting-group-free synthesis of arbusculone, andirolactone, pinnatolide, ipomolactone, cyclocapitelline and isocyclocapitelline. Tetrahedron Lett. 2018, 59, 4172–4175. [Google Scholar] [CrossRef]
- Daub, M.E.; Prudhomme, J.; Ben Mamoun, C.; Le Roch, K.G.; Vanderwal, C.D. Antimalarial properties of simplified kalihinol analogues. ACS Med. Chem. Lett. 2017, 8, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartung, J.; Drees, S.; Geiss, B.; Schmidt, P. Vanadium(V)-catalyzed oxidation of (3R)-linalool—The selective formation of furanoid linalool oxides and their conversion into isocyclocapitelline derivatives. Synlett 2003, 223–226. [Google Scholar] [CrossRef]
- Mahboobi, S.; Koller, M.; Schollmeyer, D. Synthesis of the racemates of the β-carboline alkaloid chrysotricine and its diastereomer. Monatshefte Chem. 2000, 131, 383–392. [Google Scholar] [CrossRef]
- Díez, D.; Moro, R.F.; Marcos, I.S.; Sánchez López, J.; Urones, J.G. Synthesis of an analog of usneoidol E. Part II. Synlett 2000, 794–796. [Google Scholar] [CrossRef]
- Urones, J.G.; Díez, D.; Marcos, I.S.; Basabe, P.; Lithgow, A.M.; Moro, R.F.; Garrido, N.M.; Escarcena, R. The use of acyclic monoterpenes in the preparation of β-pyrones: Synthesis of the right-hand fragment of usneoidone E. Tetrahedron 1995, 51, 3691–3704. [Google Scholar] [CrossRef]
- Serra, S.; De Simeis, D. A study on the lipase-catalysed acylation of 6,7-dihydroxy-linalool. Nat. Prod. Commun. 2016, 11, 1217–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; De Simeis, D.; Brenna, E. Lipase mediated resolution of cis- and trans-linalool oxide (pyranoid). J. Mol. Catal. B: Enzym. 2016, 133, S420–S425. [Google Scholar] [CrossRef]
- Serra, S.; De Simeis, D. Two complementary synthetic approaches to the enantiomeric forms of the chiral building block (2,6,6-trimethyltetrahydro-2H-pyran-2-yl)methanol: Application to the stereospecific preparation of the natural flavor linaloyl oxide. Catalysts 2018, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Serra, S. Bisabolane sesquiterpenes: Synthesis of (R)-(+)-sydowic acid and (R)-(+)curcumene ether. Synlett 2000, 2000, 890–892. [Google Scholar] [CrossRef]
- Klein, E.; Farnow, H.; Rojahn, W. Die chemie der linalool-oxide. Justus Liebigs Ann. Chem. 1964, 675, 73–82. [Google Scholar] [CrossRef]
- Méou, A.; Bouanah, N.; Archelas, A.; Zhang, X.M.; Guglielmetti, R.; Furstoss, R. Synthesis of all four stereoisomers of enantiomerically pure cis- and trans-linalyl oxides. Synthesis 1990, 1990, 752–753. [Google Scholar] [CrossRef]
- Méou, A.; Bouanah, N.; Archelas, A.; Zhang, X.M.; Guglielmetti, R.; Furstoss, R. Synthesis of all four stereoisomers of enantiomerically pure tetrahydro-2,2,6-trimethyl-6-vinyl-2H-pyran-3-ols. Synthesis 1991, 1991, 681–682. [Google Scholar] [CrossRef]
- Vidari, G.; Di Rosa, A.; Zanoni, G.; Bicchi, C. Enantioselective synthesis of each stereoisomer of the pyranoid linalool oxides: The linalool route. Tetrahedron Asymmetry 1999, 10, 3547–3557. [Google Scholar] [CrossRef]
- Vidari, G.; Di Rosa, A.; Castronovo, F.; Zanoni, G. Enantioselective synthesis of each stereoisomer of the pyranoid linalool oxides: The geraniol route. Tetrahedron Asymmetry 2000, 11, 981–989. [Google Scholar] [CrossRef]
- Duan, S.; Moeller, K.D. Anodic coupling reactions: Probing the stereochemistry of tetrahydrofuran formation. A short, convenient synthesis of linalool oxide. Org. Lett. 2001, 3, 2685–2688. [Google Scholar] [CrossRef]
- Volz, F.; Wadman, S.H.; Hoffmann-Röder, A.; Krause, N. Gold catalysis in stereoselective natural product synthesis: (+)-linalool oxide, (−)-isocyclocapitelline, and (−)-isochrysotricine. Tetrahedron 2009, 65, 1902–1910. [Google Scholar] [CrossRef]
- Wan, K.K.; Litz, J.P.; Vosburg, D.A. Two-step, stereoselective synthesis of linalyl oxides by asymmetric allylic o-alkylation. Tetrahedron Asymmetry 2010, 21, 2425–2428. [Google Scholar] [CrossRef]
- Al Hazmi, A.M.; Sheikh, N.S.; Bataille, C.J.R.; Al-Hadedi, A.A.M.; Watkin, S.V.; Luker, T.J.; Camp, N.P.; Brown, R.C.D. Trans-2-tritylcyclohexanol as a chiral auxiliary in permanganate-mediated oxidative cyclization of 2-methylenehept-5-enoates: Application to the synthesis of trans-(+)-linalool oxide. Org. Lett. 2014, 16, 5104–5107. [Google Scholar] [CrossRef]
- David, L.; Veschambre, H. Preparation d’oxydes de linalol par bioconversion. Tetrahedron Lett. 1984, 25, 543–546. [Google Scholar] [CrossRef]
- Miyazawa, M.; Yokote, K.; Kameoka, H. Resolution of racemic linalool oxide-pyranoid by microbial esterification. Tetrahedron Asymmetry 1995, 6, 1067–1068. [Google Scholar] [CrossRef]
- Demyttenaere, J.C.R.; Adams, A.; Vanoverschelde, J.; De Kimpe, N. Biotransformation of (S)-(+)-linalool by Aspergillus niger: An investigation of the culture conditions. J. Agric. Food Chem. 2001, 49, 5895–5901. [Google Scholar] [CrossRef] [PubMed]
- Mirata, M.-A.; Wüst, M.; Mosandl, A.; Schrader, J. Fungal biotransformation of (±)-linalool. J. Agric. Food Chem. 2008, 56, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, M.J.; Gümüs, A.; Ruijter, E.; Faber, K.; Orru, R.V.A.; Hall, M. Enantioselective bio-hydrolysis of geranyl-derived rac-epoxides: A chemoenzymatic route to trans-furanoid linalool oxide. Adv. Synth. Catal. 2019, 361, 813–825. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Entry | Experimental Conditions 1 | Conversion of Linalool Oxide (Pyranoid) % 4 | Benzoate Ester (Pyranoid) % 2,4 | Benzoate Ester (Furanoid) % 3,4 | Benzoate Esters Ratio (Pyranoid/Furanoid) |
|---|---|---|---|---|---|
| 1 | BzCl (3 eq.); DMAP cat.; rt; 4 h | >99 | 55 | 45 | 1.2 |
| 2 | BzCl (3 eq.); rt; 4 h | >99 | 73 | 27 | 2.7 |
| 3 | BzCl (1.2 eq.); from 0 °C to rt; 4 h | 96 | 97 | 3 | 32.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, S.; De Simeis, D.; Papili, S. A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide. Chemistry 2021, 3, 1247-1257. https://doi.org/10.3390/chemistry3040090
Serra S, De Simeis D, Papili S. A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide. Chemistry. 2021; 3(4):1247-1257. https://doi.org/10.3390/chemistry3040090
Chicago/Turabian StyleSerra, Stefano, Davide De Simeis, and Sara Papili. 2021. "A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide" Chemistry 3, no. 4: 1247-1257. https://doi.org/10.3390/chemistry3040090
APA StyleSerra, S., De Simeis, D., & Papili, S. (2021). A Practical Laboratory-Scale Synthesis of All Eight Stereoisomeric Forms of Terpene Linalool Oxide. Chemistry, 3(4), 1247-1257. https://doi.org/10.3390/chemistry3040090