1. Introduction
The stability of a complex between two binding partners reflects the interplay of several factors, each of which adds an individual energy contribution to the overall Gibbs free energy of complex formation Δ
G° [
1,
2]. The Δ
G° of a complex in solution can be expressed, for example, as the sum of Δ
G°
i, characterizing the intrinsic stability in the gas phase, the free desolvation energies of the individual complex constituents, and the free solvation energy of the complex [
3]. This breakdown shows that, if the Gibbs free energies required to desolvate the binding partners in a specific solvent cannot be overcompensated by the sum of the Δ
G° terms associated with the formation of the complex in the gas phase and its subsequent solvation, the overall Δ
G° will be positive and the complex will not form. Δ
G°
i itself represents the sum of the energy contributions associated with the attractive and repulsive interactions within the complex, the possible strain in the binding partners arising during complex formation, and other factors.
Separately quantifying these contributions affords energy increments that allow the comparison and classification of noncovalent interactions [
2]. Several methods exist for this purpose, one of which involves performing trend analyses in which the structure of a binding partner is systematically changed, for example by gradually increasing the number of functional groups with which the substrate interacts and quantifying the influence of this structural change on complex stability. An early application of this method by Schneider afforded an increment for the energetic contribution of salt bridges to molecular recognition processes [
4,
5,
6], and similar analyses have subsequently been performed to characterize other types of interactions [
2]. Interaction energies can also be estimated by using molecular balances [
7,
8], as demonstrated by the groups of Wilcox [
9], Diederich [
10], Cockroft [
11], and Shimizu [
12], or double-mutant cycles, which have been used extensively by Hunter and coworkers [
13].
Linear free energy relationships are also useful to analyze solvent effects on binding processes by correlating binding strength with the various parameters available to characterize solvent properties [
14]. Two recent examples from the field of anion recognition [
15,
16,
17] came in this respect from the groups of Sindelar and Johnson [
18,
19]. Sindelar demonstrated that the halide affinity of a neutral bambus[6]uril correlates with the Swain acidity parameter
A of the solvent in which complex formation was investigated [
18], and Johnson showed that the chloride affinity of a bis(arylethynyl phenylurea) host in eight different solvents correlates with the solvents’
ET(30) values [
19].
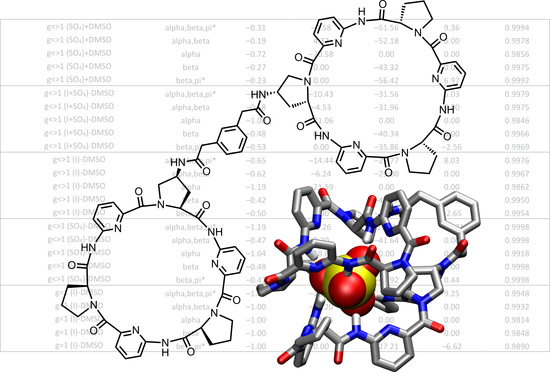
The anion affinities of bis(cyclopeptides)
1a and
1b (
Figure 1) for iodide and sulfate in various different solvents and aqueous solvent mixtures were analyzed in a similar manner [
20,
21]. These bis(cyclopeptides) belong to a family of anion receptors developed in the author’s group that contain two cyclic hexapeptide moieties with alternating proline and 6-aminopicolinic acid subunits connected covalently via one or more linkers [
22]. Each cyclopeptide ring orients the NH groups toward a shallow cavity surrounded by the proline rings. The two receptor subunits can interdigitate, thus creating a cavity in which the anion is included and where it interacts with six converging NH groups. These complexes even form in highly competitive aqueous solvent mixtures and, in the cases of
1a and
1b, even in water [
23]. Anion affinity depends on the structure of the linker [
24,
25,
26,
27,
28,
29] and the number of linking units between the two rings [
28,
29,
30].
In the cases of
1a and
1b, a statistical analysis of the Δ
G° values determined under different conditions revealed [
21] that anion affinity can be estimated by using a simple linear relationship (Equation (1)) in which the Gibbs free energy required to transfer the anion from water into the respective solvent mixture Δ
G°tr and the Kamlet–Taft parameter
α that describes the solvent’s ability to donate hydrogen bonds appear as the only variables:
Since only the two structurally related bis(cyclopeptides)
1a and
1b were considered in this analysis, the effects of receptor structure on the binding affinity were captured in the coefficients
g and
a, of which
a is formally an energy term. Other bis(cyclopeptides) can therefore be expected to yield different values for
g and
a. Alternatively,
1a and
1b can be regarded as a reference system to which the effects of the linker structure or the nature of the anion on complex stability can be related by including an additional Gibbs free energy contribution into Equation (1) that refers to the linker structure (Δ
G°
L) or the anion (Δ
G°
A) (Equation (2)):
If this relationship is valid, it should be possible to calculate ΔG°L by comparing the experimental ΔG°exp of a given bis(cyclopeptide)–anion complex with the ΔG° value estimated by using the constants g and a of the reference system and the terms ΔG°tr and α that are characteristic for the anion and the solvent in which ΔG°exp was determined. Conversely, comparing the stabilities of the complexes of different anions with the same bis(cyclopeptide) should yield ΔG°A. Importantly, since the sum g ΔG°tr + a α already encodes for the influence of the solvent on the complex stability, the increments ΔG°L and ΔG°A should be independent of the conditions under which the complex is formed.
In the following, it is shown that this treatment is indeed possible, yielding relatively consistent values of ΔG°L and ΔG°A for the bis(cyclopeptides) developed to date. While the thus determined energies only provide information about the extent to which changing the linker structure, the number of linkers, or the nature of the anion affects complex stability relative to the reference system, they can still be regarded as energy increments characteristic for the family of anion receptors for which they were determined.
3. Results
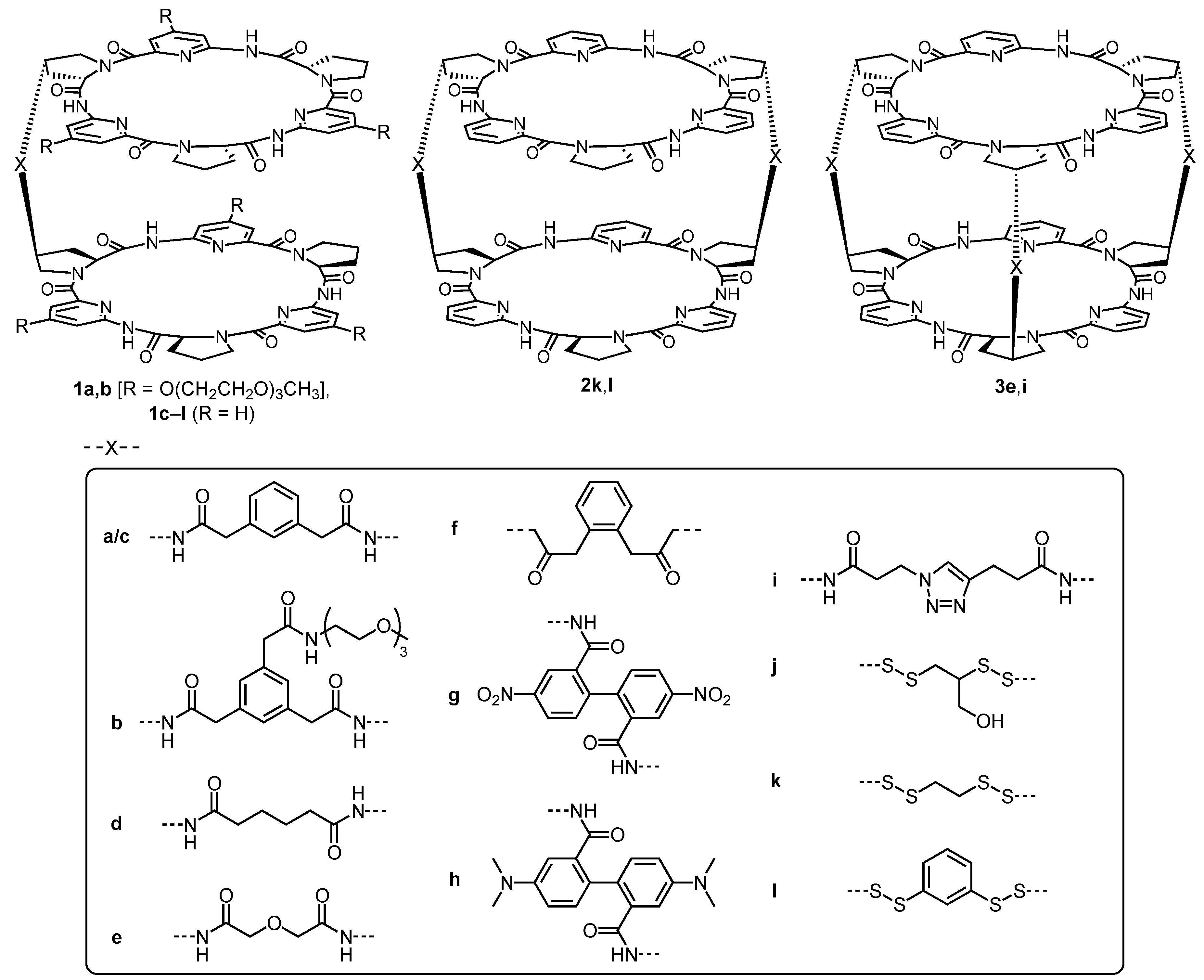
The structures of the bis(cyclopeptides) whose anion affinity was quantified in previous work are shown in
Figure 1. Compounds
1a–
l represent singly linked bis(cyclopeptides) that were either synthesized by reacting the corresponding cyclopeptide monoamine with a dicarboxylic acid [
20,
21,
24,
26,
27,
29], by connecting a cyclopeptide with an azide group with an analog containing a terminal alkyne in the presence of a copper(I) catalyst [
28], or under the conditions of disulfide exchange [
25,
30]. The doubly linked bis(cyclopeptides)
2j and
2k and triply linked receptors
3d and
3h were prepared analogously to the corresponding singly linked derivatives. The binding constants that are available for these bis(cyclopeptides) were either determined by NMR titrations or isothermal titration calorimetry. As substrates, mostly sulfate and the halides iodide, bromide, and chloride were used, and in one case nitrate affinity was also quantified. These measurements were performed in water, water/methanol, water/acetonitrile, and water/DMSO mixtures containing different fractions of the organic solvent so that a total of 121 binding constants could be used in this study. These binding constants and the corresponding free energies of complex formation Δ
G°
exp are collected in
Table S1 of the Supplementary Materials.
The choice of the reference system was based on the number of independent binding constants available for each receptor. Considering that the solubilizing substituents in the cyclopeptide and linker subunits of
1a and
1b did not markedly affect anion affinity with respect to the unsubstituted bis(cyclopeptide)
1c [
20], 60 binding constants were available for
1a–
c, of which 25 belonged to sulfate complexes and 35 to iodide complexes. Since none of the other bis(cyclopeptides) was characterized at a similar level of detail, bis(cyclopeptides)
1a–
c were chosen as reference receptors.
With 60 binding constants, the dataset used for the statistical analysis was larger than that previously considered [
21]. Moreover, the binding constants in the two subsets were analyzed simultaneously in this study, independent of the solvent in which the measurements were performed, while the earlier calculations involved analyzing the binding constants for pure solvents, water/methanol, water/acetonitrile, and water/DMSO mixtures separately. Because of the different approach chosen here, the validity of Equation (1) was reassessed by testing whether additionally considering the other Kamlet–Taft parameters
β (hydrogen bond acceptance ability) and
π* (polarizability) as variables, replacing
α with another parameter, or using Reichardt’s
ET(30) values to describe the solvent properties would improve the correlation.
For the analysis, the dimensionless solvent parameters
α,
β,
π*, and
ET(30) first had to be matched to the experimental conditions of the respective binding study. To this end, the reported values for water/methanol, water/acetonitrile, and water/DMSO mixtures [
31] were fitted to polynomials with degrees between two and seven, depending on the quality of the fit. The equations thus obtained (see
Supplementary Materials) allowed the estimation of
α,
β,
π*, and
ET(30) for each measurement after converting the solvent compositions that were previously mostly specified in terms of vol% to mol%. In a similar manner, the Gibbs free energies of transfer Δ
G°
tr were also calculated. The values for the transfer of the halides from water into water/methanol or water/acetonitrile mixtures could be taken from the literature for mixtures between 0 and 100 mol% of water [
33]. Transfer energies for sulfate from water into water/DMSO have also been reported across the whole range of solvent compositions [
32], but those of sulfate and nitrate for water/acetonitrile mixtures are not available, while they are reported for water/methanol mixtures only between 0 and 40 mol% of the organic component. Of the 121 binding constants included into
Table S1, 24 could therefore not be further considered, reducing the dataset to overall 97 binding constants. Note that the calculation of Δ
G°
tr for sulfate in water/methanol mixtures in four cases involved extrapolation to a solvent composition not reliably described by the reported Δ
G°
tr values. However, since the solvent compositions of these measurements differed not too strongly from the ones for which Δ
G°
tr values are still known, the respective binding constants were retained. All solvent parameters and Δ
G°
tr values determined in this way are included in
Table S1.
Based on these results, multiparametric analyses were performed to determine the coefficients in linear relationships that best allow predicting the stabilities of the iodide and sulfate complexes of
1a–
c. In this context, it turned out that including the binding constants of the iodide and sulfate complexes of
1a and
1b in water/DMSO mixtures and of the iodide complexes of
1a in neat organic solvents in the calculations caused a pronounced reduction in the goodness of fit. These binding constants were therefore removed from the dataset, reducing the total number of binding constants of iodide complexes to 29 and of sulfate complexes to 13.
Table 1 summarizes the coefficients obtained for this set of binding constants when fitting the experimental values of Δ
G°
exp to
g Δ
G°
tr and six different combinations of solvent parameters. In this table, the entries in each line denote the solvent parameter(s) used in the respective calculation, with
g,
e,
a,
b, and
p corresponding to the coefficients in the general equation Δ
G°
exp =
g Δ
G°
tr +
e ET(30) +
a α +
b β +
p π*. Note that all coefficients except
g formally represent energy terms.
Table 1 shows that combining Δ
G°
tr with
ET(30) did not afford a satisfactory correlation. Using the Kamlet–Taft parameters for the statistical analysis consistently led to good fits. The correlation did not significantly suffer when excluding
π*, but additionally excluding
β caused a significant drop of
R2. The fit was satisfactory, however, when only considering
β. Moreover, this fit did not substantially improve when additionally considering
π*. The fits of the linear correlations obtained for the calculations in which Δ
G°
tr was used together with
α and
β, only with
α, and only with
β are illustrated graphically in
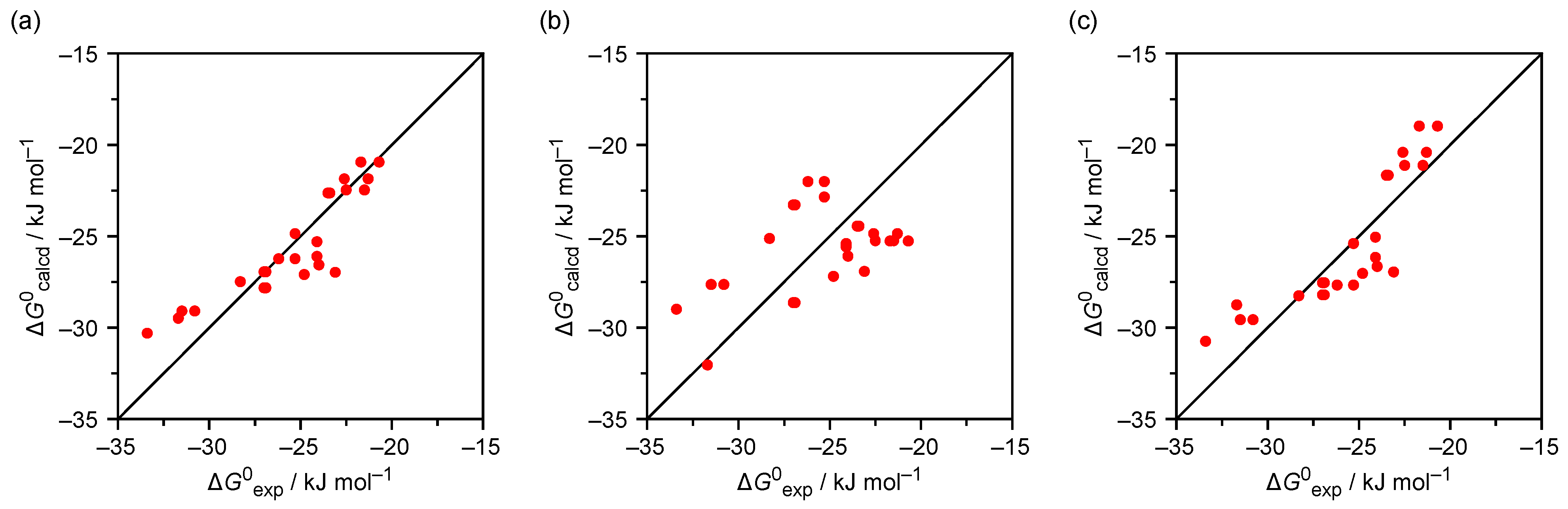
Figure 2.
In the case of the sulfate complexes, all calculations in which Kamlet–Taft parameters were considered had very good fits, with
β alone again affording a better fit than
α alone (
Table 2). The respective graphs are depicted in the
Supplementary Materials.
Note that the coefficients
g and
a in entries 4 of
Table 1 and
Table 2 are similar to those determined in the earlier study [
33] and that some calculations afforded comparable coefficients for the iodide and the sulfate complexes (e.g.,
g and
a in entries 4, or
g and
b in entries 5), indicating that the intrinsic affinity of bis(cyclopeptides)
1a–
c for both anions is likely similar.
These calculations thus showed that Equation (1) may not be optimal for estimating the experimental binding energies. The correlations were significantly better both for the iodide and the sulfate complexes of
1a–
c when additionally including
β into the equation and neglecting
α entirely did not even have a large impact on the goodness of fit. Accordingly, the calculation of Δ
G°
calcd for all the bis(cyclopeptide) complexes considered in this study was primarily based on Equation (3). In some cases, the effects of neglecting either
a or
β on the outcome of the calculations were additionally assessed.
In the next step, the consistency of the obtained results was evaluated by calculating the Gibbs free energies of the sulfate and iodide complexes of
1a–
c with the coefficients obtained in the multiparametric analyses and comparing them with the corresponding experimental free energies by calculating the difference Δ
G°
rel = Δ
G°
exp − (
g Δ
G°
tr +
a α +
b β). When using the coefficients
g,
a, and
b for the iodide complexes, the calculated Gibbs free energies matched the 29 experimental ones rather well, with only three calculated values deviating from the corresponding experimental ones by more than 2.5 kJ mol
−1 (
Figure 2a), resulting in a standard deviation of ±1.5 kJ mol
−1 (
Table 3). All calculated Gibbs free energies of the 13 sulfate complexes matched the experimental ones within an error margin of ±2.1 kJ mol
−1 (standard deviation ± 0.9 kJ mol
−1). When using the coefficients determined for the sulfate complex instead, the standard deviation of the experimental and calculated Gibbs free energies of iodide complexation amounted to ±1.9 kJ mol
−1, with five calculated values differing from the experimental ones by more than 2.5 kJ mol
−1. The same coefficients allowed predicting the stabilities of the sulfate complexes very well, with a standard deviation of ±0.4 kJ mol
−1. Thus, although the coefficients in entries 3 of
Table 1 and
Table 2 differ substantially, each combination provided relatively reliable estimates of the stabilities of the respective other complexes, with the coefficients obtained for the iodide complexes working somewhat better. Almost the same correlations were observed when omitting the term for
α from Equation (3), again demonstrating that
β is the more important solvent parameter to predict complex stability. The quality of the results was significantly lower when only considering α, although the coefficients
g and
a in entries 4 of
Table 1 and
Table 2 are comparable.
Having thus established that the coefficients obtained in the statistical analysis for the iodide and sulfate complexes of
1a–
c allow a relatively reliable prediction of complex stability for a wide variety of solvent mixtures, the calculations were extended to the anion complexes of the other bis(cyclopeptides). To this end, Gibbs free energies were calculated for each bis(cyclopeptide)–anion combination by using the coefficients
g,
a, and
b in entries 3 of
Table 1 and
Table 2 and the values of Δ
G°
tr,
α, and
β corresponding to the conditions of the respective measurements. The results obtained were subtracted from the corresponding experimental Gibbs free energies of complex formation (Δ
G°
rel = Δ
G°
exp − Δ
G°
calcd) to assess the extent to which the experimental binding energies differ from those expected for iodide or sulfate complexes of a bis(cyclopeptides)
1a–
c under the same conditions. The Δ
G°
rel values calculated in this way are summarized in
Table 3. If more than one binding constant was available for a specific bis(cyclopeptide)–anion combination, the values in the table represent averages of the specified number of binding constants.
Once values for Δ
G°
rel were available, the energy increments Δ
G°
A and Δ
G°
L could be calculated. Δ
G°
A describes the difference in binding energies resulting from exchanging the anion of the reference system for another anion. These increments can thus be calculated by subtracting the Δ
G°
rel value in
Table 3 for a given bis(cyclopeptide)–anion pair from the Δ
G°
rel value of the iodide complex of the same bis(cyclopeptide). The Δ
G°
rel values calculated in this way are summarized in
Table 4. Values in parentheses in this table were calculated by using the coefficients
g,
a, and
b obtained for the sulfate complexes of bis(cyclopeptides)
1a–
c.
While the results should be most reliable for the bis(cyclopeptides) for which the statistical analysis was performed, the other bis(cyclopeptides) should afford comparable Δ
G°
rel values for a given anion.
Table 4 shows that this is mostly the case, with a few exceptions. The absolute increments obtained for the sulfate complex of
1g and the chloride complex of
1f are larger than the respective other increments, for example. The averages of the entries in the columns of
Table 4 nevertheless illustrate how the stability of a complex responds when iodide is exchanged for another anion. Positive increments indicate that the calculated Gibbs free energy of complex formation is more negative than the experimental one, and that the respective complex is therefore less stable than the reference system. Accordingly, the increments in
Table 4 suggest that the iodide and sulfate complexes of the bis(cyclopeptides) are indeed intrinsically comparably stable. Bromide is bound less strongly than iodide or sulfate, and the least stable complexes are formed with chloride and nitrate.
The linker increments Δ
G°
L can be calculated in a similar manner by relating the Δ
G°
rel values of bis(cyclopeptides)
1a–
c with the corresponding values of the other bis(cyclopeptides). These increments are summarized in
Table 5, in which rows are now expected to contain comparable values because the influence of the linker on the bis(cyclopeptide) complexes should be similar, independent of the anion (the Δ
G°
L value for the nitrate complex of
1d is missing because the stabilities of the nitrate complexes of
1a–
c are unknown). The comparison of the values in rows containing more than one entry shows that this is again mostly the case. The averages calculated for Δ
G°
L within a row thus allow estimating the effects of the linker on complex stability. Accordingly, the doubly linked bis(cyclopeptide)
2k has the intrinsically highest anion affinity, which is indeed the case [
30], while the presence of three linkers in bis(cyclopeptides)
3e and
3i does not seem to be very beneficial.
4. Discussion
The multiparametric analyses of the binding energies of 29 iodide complexes and 13 sulfate complexes of bis(cyclopeptides) 1a–c demonstrated that anion affinity can be predicted rather reliably on the basis of a few parameters in a wide range of solvent mixtures. The most important parameter is the Gibbs free energy ΔG°tr of transferring an anion from water into the respective solvent mixture. These transfer energies are mostly positive, while g is always negative, indicating that complex stability increases with the increasing energetic cost of transferring an anion into a solvent mixture. In other words, the less favorable the anion solvation in a solvent mixture in terms of the Gibbs free energy, the greater the energetic gain of complex formation. Interestingly, the multiparametric analyses afforded coefficients for ΔG°tr close to 1 in a few cases, suggesting that ΔG°tr contributes exactly once to anion binding. However, fixing the coefficient g to 1 mostly afforded unsatisfactory correlations, particularly when including the solvent parameter β in the analysis (data not shown), which is why g was also fitted.
Besides Δ
G°
tr, additional solvent parameters have to be considered in the linear correlation to predict binding strength. As observed earlier, the Kamlet–Taft parameters
α, describing the hydrogen bond acidity of the solvent mixture, and
β, relating to the corresponding basicity, afforded better correlations than Reichardt’s
ET(30) parameter [
21]. Moreover, including only
α in the linear equation consistently yielded poorer correlations than additionally or even exclusively including
β. This correlation differs from that found in previous work, where
α was considered more important, which could have several reasons. A possible but probably not decisive one is the larger dataset used in this work. More important is likely that Gibbs free energies were included into the previous analysis that were obtained for iodide complexes in methanol, acetonitrile, and DMSO. These binding energies were not considered here because they consistently caused a reduction in the quality of the fit. The earlier work also contained binding energies for complex formation in water/DMSO mixtures, which turned out to be difficult to evaluate together with the results obtained in the other solvents. Possible reasons could be that Δ
G°
tr increases when going from water to water/DMSO mixtures but iodide affinity decreases, which is opposite to the trends in the other solvents. In addition, the transfer energies of sulfate into water/DMSO mixtures are comparably large, which causes small changes in the solvent composition to have large effects on the calculated binding energies [
32]. As a consequence, calculations with coefficients obtained when considering the binding energies obtained in water/DMSO mixtures afforded increments with relatively large errors that were, however, comparable in magnitude to those in
Table 4 and
Table 5.
For the purpose of this work, the exact choice of parameters used to correlate the experimental and calculated binding energies is secondary, as long as a parameter combination exists that allows predicting binding strengths. The question is nevertheless justified, why
α and especially
β worked so well. Both the hydrogen bond donor and acceptor strength of the solvent should indeed affect anion binding since increasing the donor strength should improve anion solvation and increasing the acceptor strength should improve the solvation of the receptor donors that interact with the anion. Accordingly, one should expect anion binding to become stronger as the values of
α and
β decrease and the solvent becomes less competitive. The negative signs of most coefficients
a and
b in the entries 3, 4, and 5 of
Table 1 and
Table 2 demonstrate, however, that the opposite is generally the case (
α and
β are usually positive). The observed trends can therefore not be rationalized in a straightforward manner. In the previous work, the dependence on
α was tentatively attributed to the contribution of the hydrophobic effect to anion binding since
α is larger in water than in solvent mixtures [
21]. The parameter
β typically exhibits the opposite trend, mirroring to some extent the increase in complex stability with decreasing water content of the solution, but the transfer energies Δ
G°
tr are likely more important in this context. Accordingly, understanding the correlation of anion affinity with
α and
β requires further work.
Independent of this aspect, the calculations performed in this study afforded relatively consistent energy increments, describing the effects of changing the anion or the receptor structure on binding strength. One major drawback is that the dataset was too small to obtain statistically meaningful increments for all the bis(cyclopeptide)–anion combinations studied to date. The obtained increments nevertheless correctly reflect previously observed trends as illustrated by the selection of binding energies measured in water/methanol mixtures in
Table 6.
The correlation between experimental and calculated Δ
G°
L is somewhat poorer for binding energies measured in water/acetonitrile mixtures as shown by the results in
Table 7. In particular, the positive influence of the disulfide-containing linkers on complex stability is underestimated in the calculated increments, maybe because these linkers differ structurally too strongly from the amide-based ones in the other bis(cyclopeptides). That connecting the cyclopeptide rings with more than one linker is often beneficial is, however, correctly predicted by the Δ
G°
L values in
Table 5.
The validation of the ΔG°A values for different anions has to be based on binding energies measured in water to eliminate the effect of the transfer energies that differ from anion to anion. The corresponding binding studies with 1a and 1b yielded experimental ΔG°A values for sulfate, bromide, and chloride complexation with respect to the stability of the iodide complex amounting to 1.7, 2.2 and 8.3 kJ mol−1, respectively, which match the corresponding calculated values of 0.6 (−0.5), 3.4 (2.9), and 8.5 (8.0) reasonably well. Accordingly, the correlation between experimental and calculated energy increments derived in this study is acceptable, especially when considering that only a limited number of binding constants was considered. Based on these increments, the effects of receptor structure and nature of the anion on complex stability in relation to the reference system can now be quantified.






{kind=link}
{kind=link}
{kind=link}