Insights from Initial Variant Detection by Sequencing Single Sperm in Cattle

 , , , and
, , , and
Abstract
:1. Background
2. Methods
2.1. Sample Collection and Whole Genome Amplification and Sequencing
2.2. Genotype Calling
2.3. Filtration of SNPs, INDELs, and Samples
2.4. De Novo Mutation Detection
2.5. Gene Annotation
2.6. Amplification and Sequencing of Cattle Mutations
3. Results
3.1. Sequencing and Genotyping of Haploid Sperms
3.2. Sequencing and Genotyping of Sample1 Diploid Somatic Genome
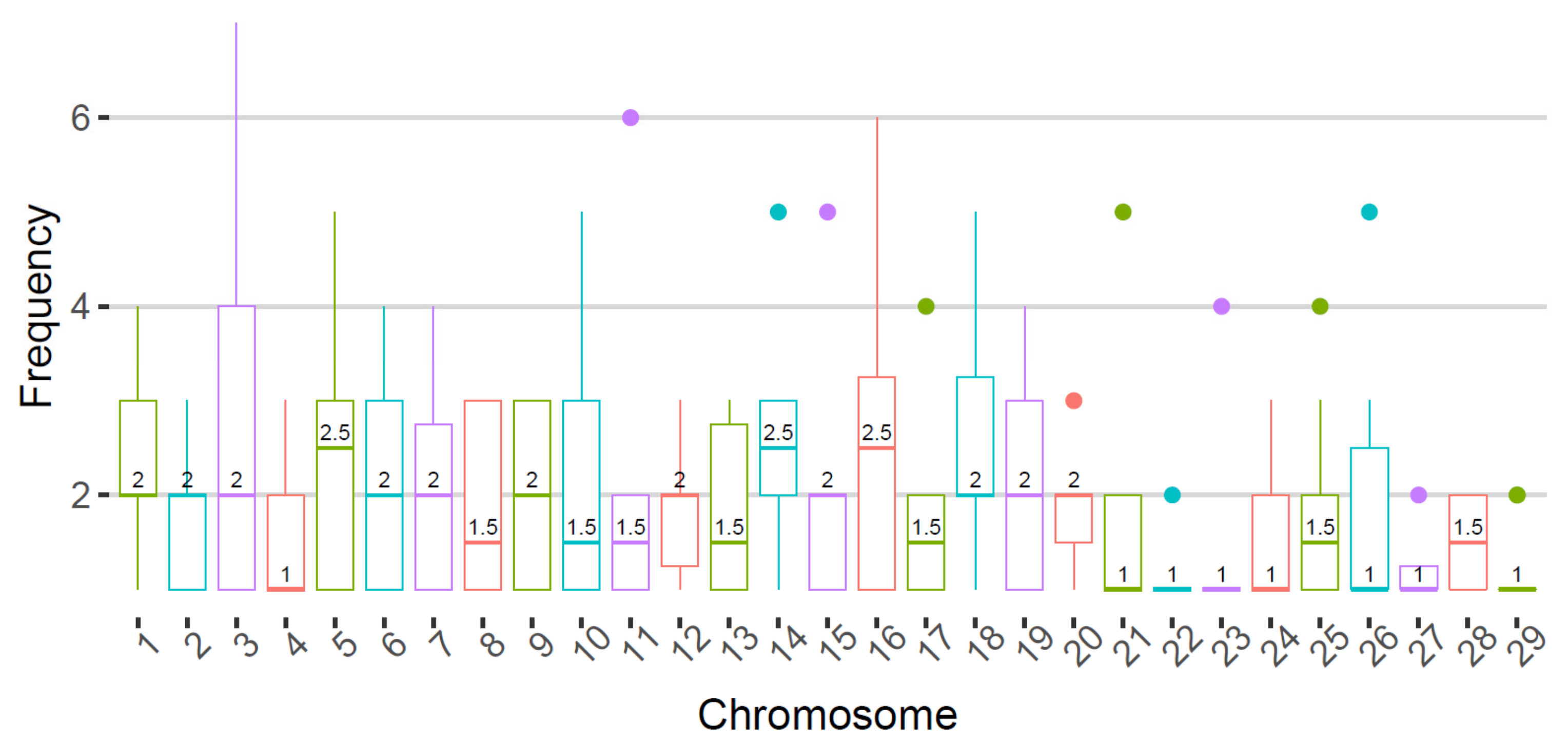
3.3. De Novo Mutations Detected in Single Sperms
3.4. PCR Sanger Sequencing Validation of De Novo Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
Abbreviations
| AGIL | Animal Genomics and Improvement Laboratory |
| BQSR | base quality score recalibration |
| CDS | coding sequence |
| DNM | de novo mutations |
| DPR | Daughter pregnancy rate |
| FS | Fisher strand |
| INDEL | insertion and deletion |
| Kb | kilobase pairs |
| MALBAC | multiple annealing and looping based amplification cycles |
| Mb | megabase pairs |
| MQ | mapping quality |
| PCR | polymerase chain reaction |
| PGC | primordial germ cell |
| QC | quality control |
| QD | quality by depth |
| QUAL | quality score |
| scDNA-seq | single-cell DNA-seq |
| SNP | single nucleotide polymorphism |
| SNV | single nucleotide variation |
| WGA | whole-genome amplification |
References
- Wang, J.; Fan, H.C.; Behr, B.; Quake, S.R. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 2012, 150, 402–412. [Google Scholar]
- Lu, S.; Zong, C.; Fan, W.; Yang, M.; Li, J.; Chapman, A.R.; Zhu, P.; Hu, X.; Xu, L.; Yan, L.; et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 2012, 338, 1627–1630. [Google Scholar]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar]
- Smith, G.P. Evolution of repeated DNA sequences by unequal crossover. Science 1976, 191, 528–535. [Google Scholar]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar]
- Bell, A.D.; Mello, C.J.; Nemesh, J.; Brumbaugh, S.A.; Wysoker, A.; McCarroll, S.A. Insights into variation in meiosis from 31,228 human sperm genomes. Nature 2020, 583, 259–264. [Google Scholar]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; MacKenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar]
- Zhou, Y.; Shen, B.; Jiang, J.; Padhi, A.; Park, K.E.; Oswalt, A.; Sattler, C.G.; Telugu, B.P.; Chen, H.; Cole, J.B.; et al. Construction of PRDM9 allele-specific recombination maps in cattle using large-scale pedigree analysis and genome-wide single sperm genomics. DNA Res. 2018, 25, 183–194. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar]
- Hou, Y.; Fan, W.; Yan, L.; Li, R.; Lian, Y.; Huang, J.; Li, J.; Xu, L.; Tang, F.; Xie, X.S.; et al. Genome analyses of single human oocytes. Cell 2013, 155, 1492–1506. [Google Scholar]
- Zafar, H.; Wang, Y.; Nakhleh, L.; Navin, N.; Chen, K. Monovar: Single-nucleotide variant detection in single cells. Nat. Methods 2016, 13, 505–507. [Google Scholar]
- Dong, X.; Zhang, L.; Milholland, B.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods 2017, 14, 491–493. [Google Scholar]
- Luquette, L.J.; Bohrson, C.L.; Sherman, M.A.; Park, P.J. Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat. Commun. 2019, 10, 3908. [Google Scholar]
- Conrad, D.F.; Bird, C.; Blackburne, B.; Lindsay, S.; Mamanova, L.; Lee, C.; Turner, D.J.; Hurles, M.E. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat. Genet. 2010, 42, 385–391. [Google Scholar]
- Makova, K.D.; Li, W.H. Strong male-driven evolution of DNA sequences in humans and apes. Nature 2002, 416, 624–626. [Google Scholar]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016, 17, 241. [Google Scholar]
- Breuss, M.W.; Antaki, D.; George, R.D.; Kleiber, M.; James, K.N.; Ball, L.L.; Hong, O.; Mitra, I.; Yang, X.; Wirth, S.A.; et al. Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat. Med. 2020, 26, 143–150. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Sperm ID | 12 | 32 | 33 | 34 | 36 | 43 | 71 | 76 | 88 | 92 |
|---|---|---|---|---|---|---|---|---|---|---|
| Physical coverage (Gbp) | 11.34 | 12.39 | 29.30 | 24.05 | 20.56 | 22.00 | 19.46 | 18.32 | 18.64 | 12.25 |
| Mutation counts | 34 | 39 | 38 | 35 | 27 | 28 | 49 | 57 | 94 | 45 |
| Mutation rates (×10−8) | 1.37 | 1.57 | 1.53 | 1.41 | 1.08 | 1.12 | 1.97 | 2.29 | 3.78 | 1.81 |
| Transition/Transversion ratio | 2.78 | 1.60 | 2.80 | 1.50 | 1.08 | 1.33 | 2.50 | 2.00 | 3.27 | 3.09 |
| CpG | 0.00 | 0.00 | 0.03 | 0.06 | 0.00 | 0.00 | 0.00 | 0.04 | 0.04 | 0.02 |
| Coding-missense | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 2 | 2 | 2 |
| UTR | 0 | 1 | 0 | 1 | 0 | 3 | 2 | 0 | 2 | 1 |
| Noncoding genes | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| Pseudo genes | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Protein coding genes | 12 | 12 | 14 | 19 | 11 | 19 | 16 | 26 | 44 | 19 |
| Exonic | 0 | 1 | 1 | 1 | 0 | 1 | 2 | 2 | 3 | 4 |
| CDS | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 2 | 2 | 2 |
| Intronic | 12 | 10 | 13 | 17 | 11 | 15 | 12 | 24 | 39 | 14 |
| Intergenic | 22 | 27 | 24 | 16 | 15 | 9 | 32 | 30 | 49 | 25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Gao, Y.; Boschiero, C.; Li, L.; Zhang, H.; Ma, L.; Liu, G.E. Insights from Initial Variant Detection by Sequencing Single Sperm in Cattle. Dairy 2021, 2, 649-657. https://doi.org/10.3390/dairy2040050
Yang L, Gao Y, Boschiero C, Li L, Zhang H, Ma L, Liu GE. Insights from Initial Variant Detection by Sequencing Single Sperm in Cattle. Dairy. 2021; 2(4):649-657. https://doi.org/10.3390/dairy2040050
Chicago/Turabian StyleYang, Liu, Yahui Gao, Clarissa Boschiero, Li Li, Hongping Zhang, Li Ma, and George E. Liu. 2021. "Insights from Initial Variant Detection by Sequencing Single Sperm in Cattle" Dairy 2, no. 4: 649-657. https://doi.org/10.3390/dairy2040050
APA StyleYang, L., Gao, Y., Boschiero, C., Li, L., Zhang, H., Ma, L., & Liu, G. E. (2021). Insights from Initial Variant Detection by Sequencing Single Sperm in Cattle. Dairy, 2(4), 649-657. https://doi.org/10.3390/dairy2040050