

The Role of Ketone Bodies in Various Animal Models of Kidney Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
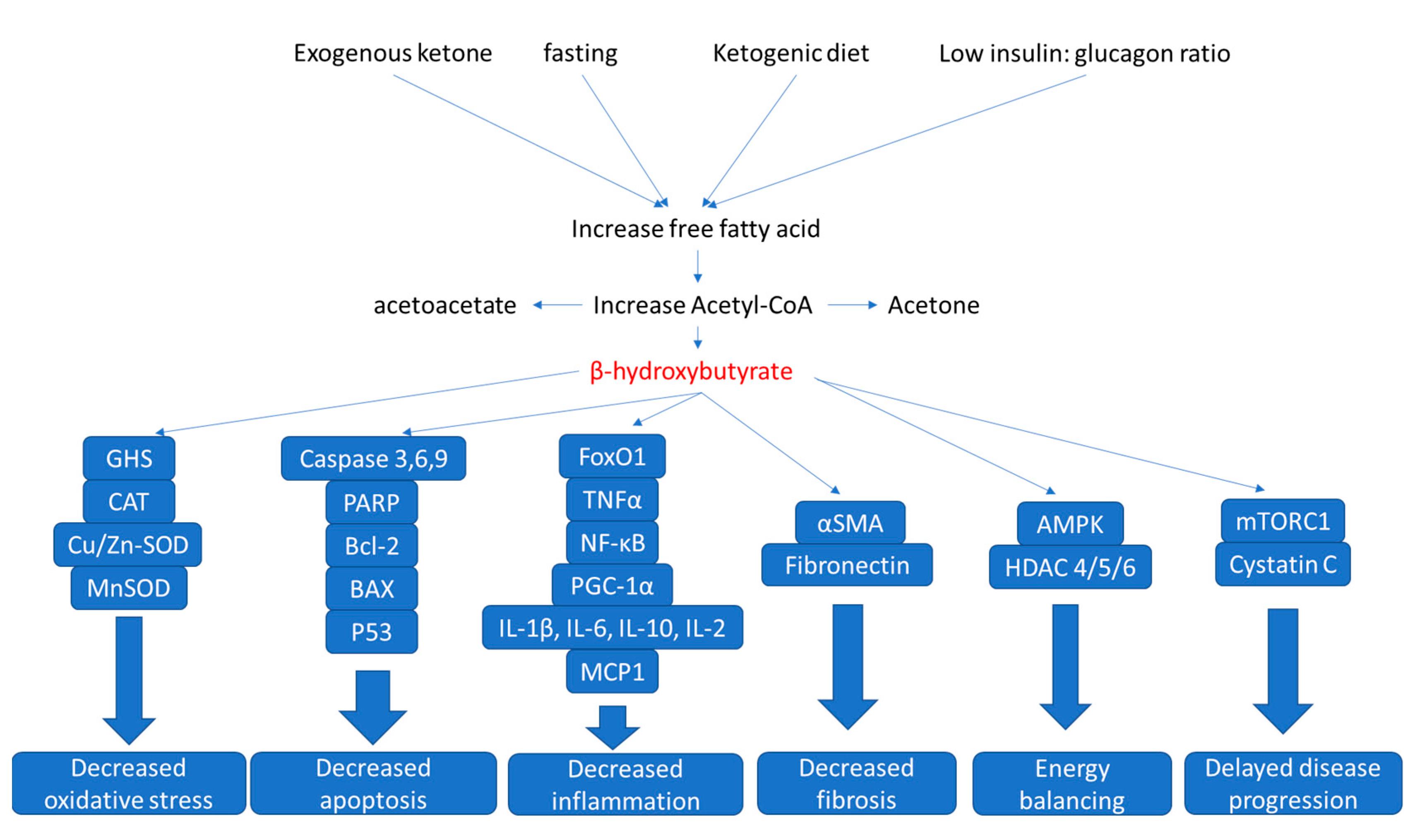
:1. Kidney Functions and Diseases
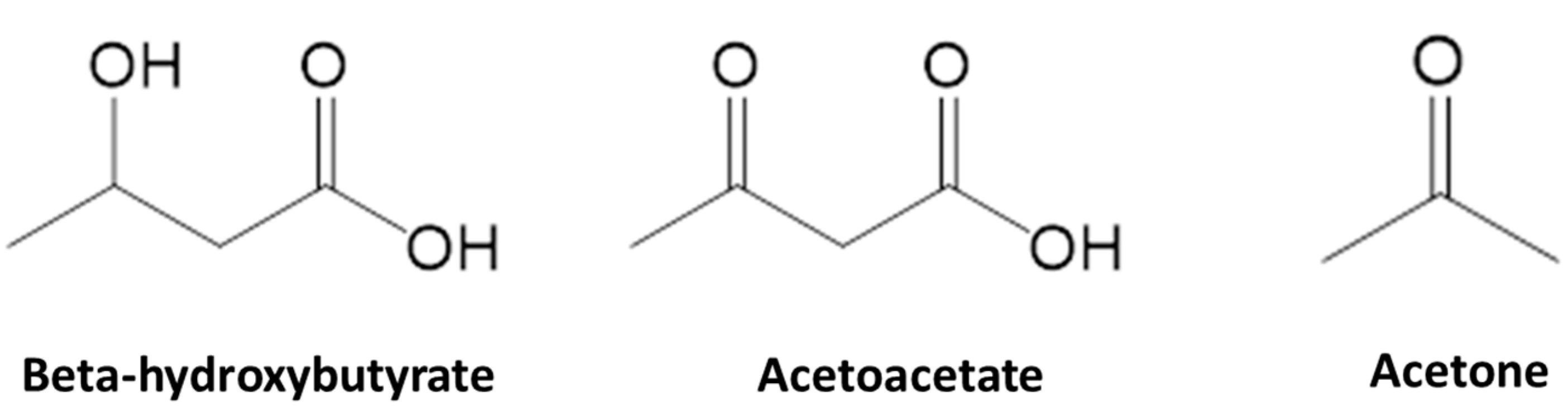
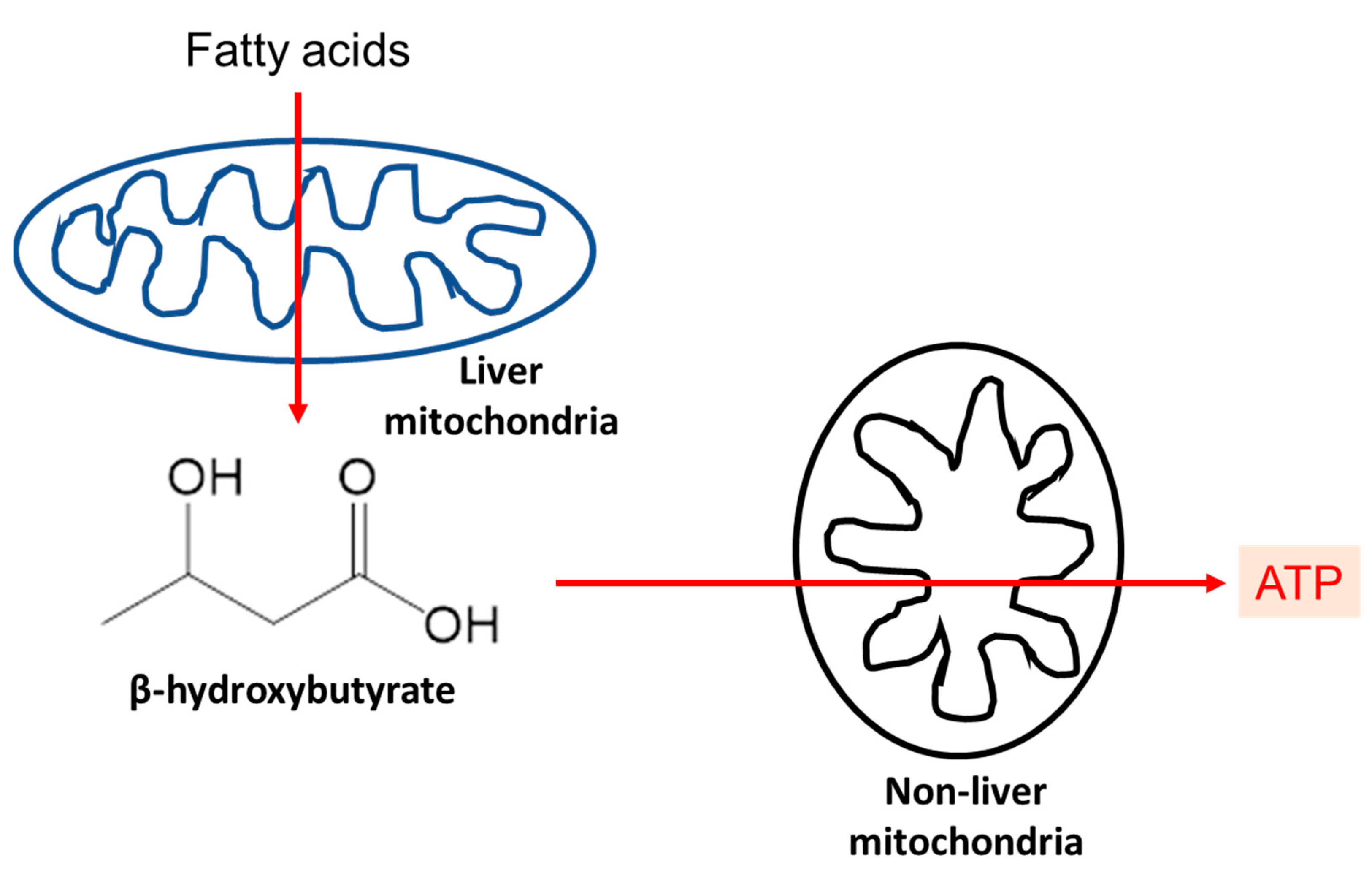
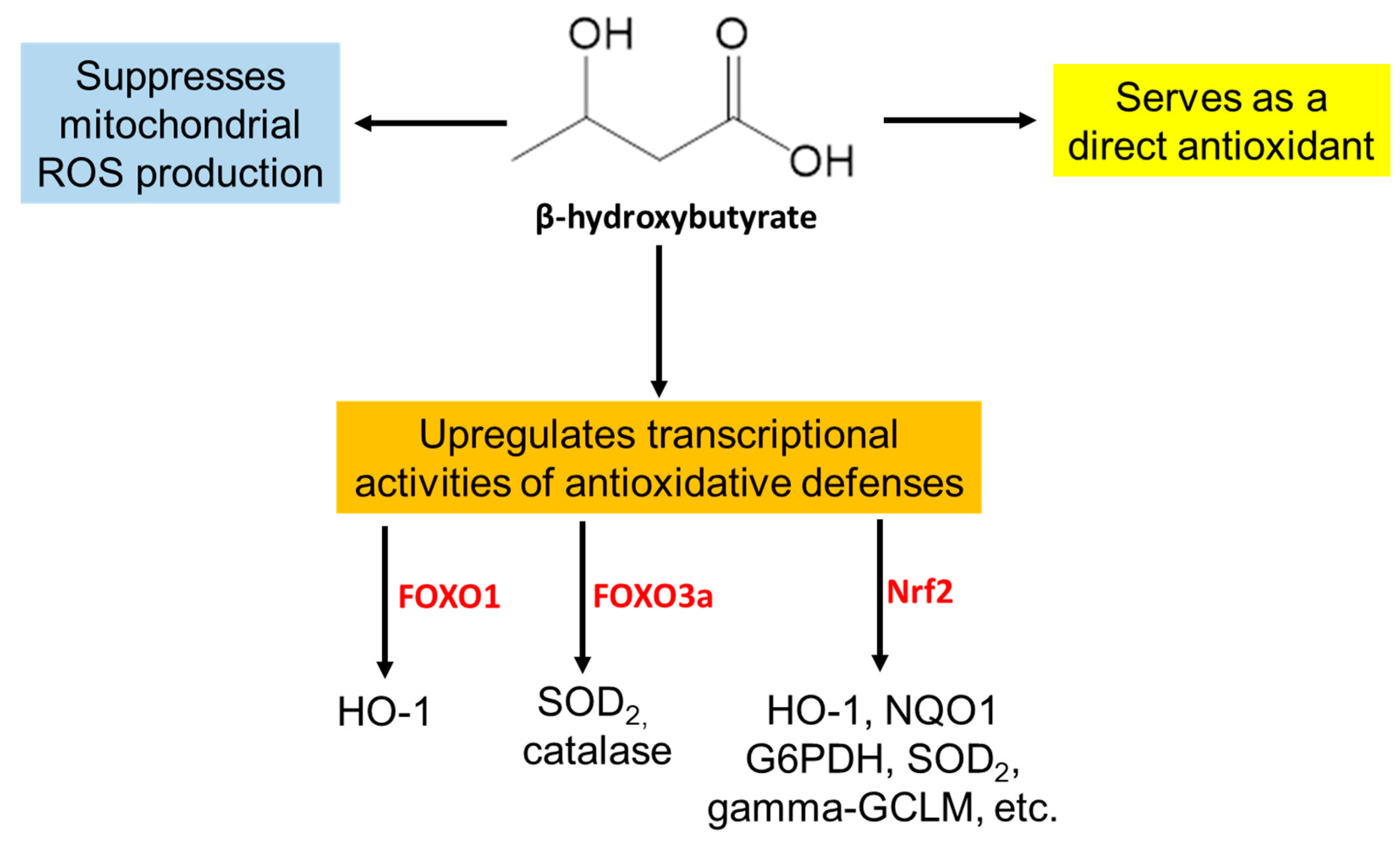
2. Ketone Bodies
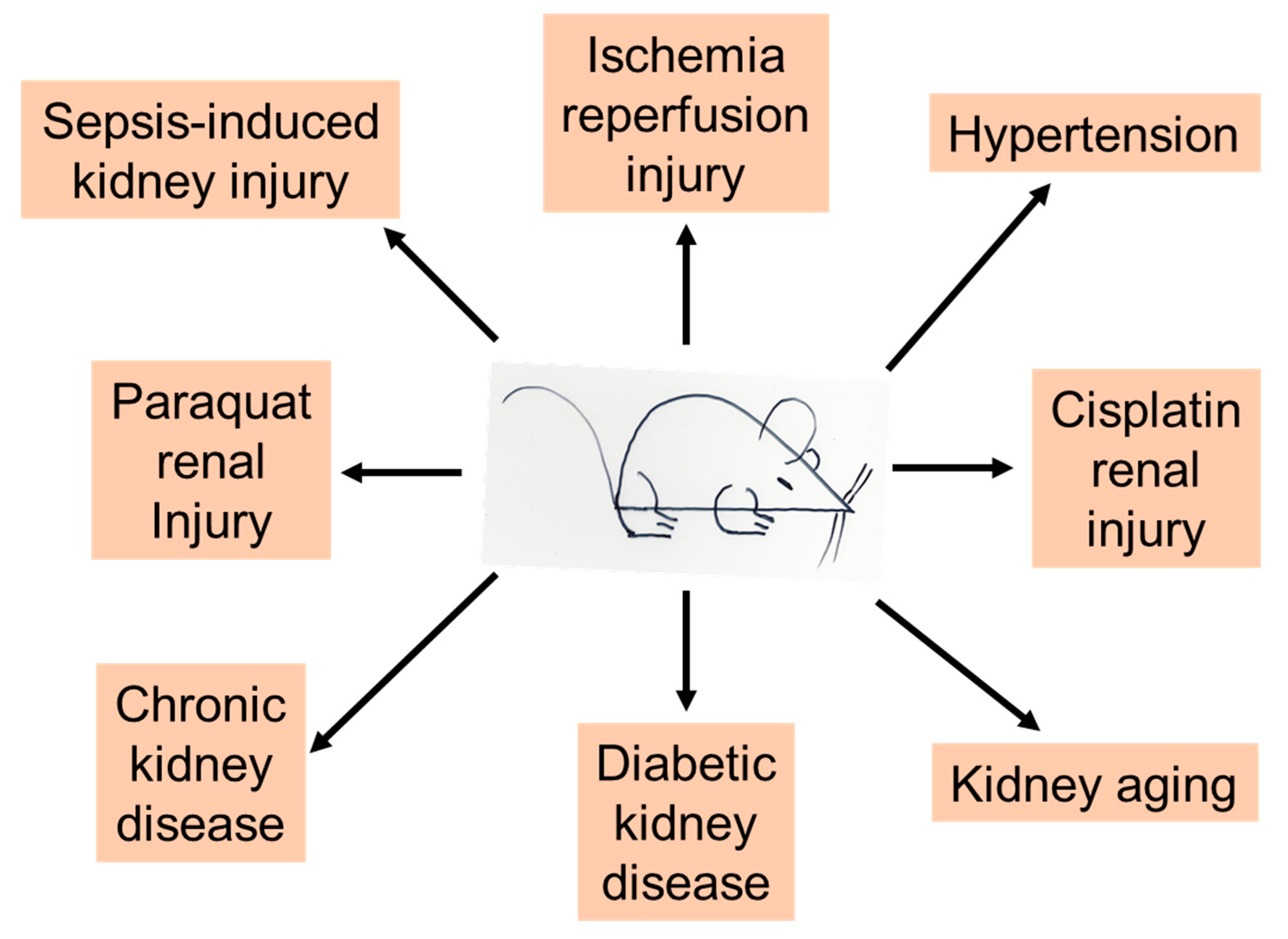
2.1. Ketone Bodies in Sepsis-Induced AKI
2.2. Ketone Bodies in Paraquat-Induced AKI
2.3. Ketone Bodies in Cisplatin-Induced AKI
2.4. Ketone Bodies in Ischemic Kidney Injury
2.5. Ketone Bodies in Hypertension and CKD Progression
2.6. Ketone Bodies in Diabetic Kidney Disease
2.6.1. SGLT2 Inhibitor (SGLT2i) and Ketones
2.6.2. Ketone Bodies and Glomerular Podocyte Senescence
2.7. Ketone Bodies in Kidney Aging
3. Summary
4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Acharya, V.; Olivero, J. The kidney as an endocrine organ. Methodist Debakey Cardiovasc. J. 2018, 14, 305–307. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar]
- van der Kooij, M.A.; Groenendaal, F.; Kavelaars, A.; Heijnen, C.J.; van Bel, F. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res. Rev. 2008, 59, 22–33. [Google Scholar] [CrossRef]
- Wu, J.; Luo, X.; Thangthaeng, N.; Sumien, N.; Chen, Z.; Rutledge, M.A.; Jing, S.; Forster, M.J.; Yan, L.J. Pancreatic mitochondrial complex i exhibits aberrant hyperactivity in diabetes. Biochem. Biophys. Rep. 2017, 11, 119–129. [Google Scholar] [CrossRef]
- Diwan, V.; Brown, L.; Gobe, G.C. Adenine-induced chronic kidney disease in rats. Nephrology 2018, 23, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.J. Folic acid-induced animal model of kidney disease. Animal Model. Exp. Med. 2021, 4, 329–342. [Google Scholar] [CrossRef]
- Yan, L.J. Nadh/nad(+) redox imbalance and diabetic kidney disease. Biomolecules 2021, 11, 730. [Google Scholar] [CrossRef]
- Turgut, F.; Awad, A.S.; Abdel-Rahman, E.M. Acute kidney injury: Medical causes and pathogenesis. J. Clin. Med. 2023, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; James, M.T. Acute kidney injury. Ann. Intern. Med. 2018, 168, 837. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.S.; Kaushik, R.; Ahmad, S.; Kaushik, R.M. Profile of acute kidney injury in patients with decompensated cirrhosis at a tertiary-care center in uttarakhand, india. Dig. Dis. 2020, 38, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, M.; Nayfeh, T.; Atieh, A.; AbuShamma, O.; Babaa, B.; Baniowda, M.; Hrizat, A.; Hasan, B.; Hassett, L.; Hamadah, A.; et al. Diagnostic performance of fractional excretion of sodium for the differential diagnosis of acute kidney injury: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2022, 17, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Dipiro, J.T.; Talbet, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M. Pharmacotherapy: A Pathophysiological Approach, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2014. [Google Scholar]
- Mulay, S.R.; Evan, A.; Anders, H.J. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol. Dial. Transplant. 2014, 29, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Jha, R.K.; Keerti, A. Chronic kidney disease: Its relationship with obesity. Cureus 2022, 14, e30535. [Google Scholar] [CrossRef]
- Yan, L.J. The nicotinamide/streptozotocin rodent model of type 2 diabetes: Renal pathophysiology and redox imbalance features. Biomolecules 2022, 12, 1225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Y.; Zhao, X.; Cui, H.; Han, M.; Ren, X.; Gang, X.; Wang, G. Obesity and chronic kidney disease. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E24–E41. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Levin, A.; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Ammirati, A.L. Chronic kidney disease. Rev. Assoc. Med. Bras. (1992) 2020, 66 (Suppl. S1), s03–s09. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in aki to ckd progression. Am. J. Physiol. Renal. Physiol. 2018, 315, F1501–F1512. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies, stress response, and redox homeostasis. Redox. Biol. 2020, 29, 101395. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Rohling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Lieberman, M.; Marks, A.D. Marks’ Basic Medical Biochemistry: A Clinical Approach, 4th ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Ghimire, P.; Dhamoon, A.S. Ketoacidosis. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fukao, T.; Mitchell, G.; Sass, J.O.; Hori, T.; Orii, K.; Aoyama, Y. Ketone body metabolism and its defects. J. Inherit. Metab. Dis. 2014, 37, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies for kidney injury and disease. Adv. Redox Res. 2021, 2, 100009. [Google Scholar] [CrossRef]
- Yan, L.J.; Allen, D.C. Cadmium-induced kidney injury: Oxidative damage as a unifying mechanism. Biomolecules 2021, 11, 1575. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. Sglt2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mtorc1 inhibition. Cell Metab. 2020, 32, 404–419 e406. [Google Scholar] [CrossRef]
- Ishimwe, J.A.; Garrett, M.R.; Sasser, J.M. 1,3-butanediol attenuates hypertension and suppresses kidney injury in female rats. Am. J. Physiol. Renal. Physiol. 2020, 319, F106–F114. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, M.H.; Ha, S.; Bang, E.J.; Lee, Y.; Lee, A.K.; Lee, J.; Yu, B.P.; Chung, H.Y. Anti-inflammatory action of beta-hydroxybutyrate via modulation of pgc-1alpha and foxo1, mimicking calorie restriction. Aging 2019, 11, 1283–1304. [Google Scholar] [CrossRef]
- Mikami, D.; Kobayashi, M.; Uwada, J.; Yazawa, T.; Kamiyama, K.; Nishimori, K.; Nishikawa, Y.; Morikawa, Y.; Yokoi, S.; Takahashi, N.; et al. Beta-hydroxybutyrate, a ketone body, reduces the cytotoxic effect of cisplatin via activation of hdac5 in human renal cortical epithelial cells. Life Sci. 2019, 222, 125–132. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Leon-Contreras, J.C.; Sanchez-Tapia, M.; Silva-Palacios, A.; Cano-Martinez, A.; Gonzalez-Reyes, S.; Jimenez-Osorio, A.S.; Hernandez-Pando, R.; Osorio-Alonso, H.; Sanchez-Lozada, L.G.; et al. A ketogenic diet attenuates acute and chronic ischemic kidney injury and reduces markers of oxidative stress and inflammation. Life Sci. 2022, 289, 120227. [Google Scholar] [CrossRef]
- Wei, T.; Tian, W.; Liu, F.; Xie, G. Protective effects of exogenous beta-hydroxybutyrate on paraquat toxicity in rat kidney. Biochem. Biophys. Res. Commun. 2014, 447, 666–671. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [Green Version]
- Thai, P.N.; Miller, C.V.; King, M.T.; Schaefer, S.; Veech, R.L.; Chiamvimonvat, N.; Bers, D.M.; Dedkova, E.N. Ketone ester d-beta-hydroxybutyrate-(r)-1,3 butanediol prevents decline in cardiac function in type 2 diabetic mice. J. Am. Heart Assoc. 2021, 10, e020729. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Kumar, S.; Behl, T.; Sachdeva, M.; Sehgal, A.; Kumari, S.; Kumar, A.; Kaur, G.; Yadav, H.N.; Bungau, S. Implicating the effect of ketogenic diet as a preventive measure to obesity and diabetes mellitus. Life Sci. 2021, 264, 118661. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef]
- Doi, K.; Leelahavanichkul, A.; Yuen, P.S.; Star, R.A. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Investig. 2009, 119, 2868–2878. [Google Scholar] [CrossRef] [Green Version]
- Kiyonaga, N.; Moriyama, T.; Kanmura, Y. Effects of dexmedetomidine on lipopolysaccharide-induced acute kidney injury in rats and mitochondrial function in cell culture. Biomed. Pharmacother. 2020, 125, 109912. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Cha, D.R.; Kim, B.; An, E.J.; Lee, S.R.; Cha, J.J.; Kang, Y.S.; Ghee, J.Y.; Han, J.Y.; Bae, Y.S. Lps-induced acute kidney injury is mediated by nox4-sh3yl1. Cell Rep. 2020, 33, 108245. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Song, Y.; Zhao, M.; Yi, Z.; Zeng, Q. Protective effects of edaravone, a free radical scavenger, on lipopolysaccharide-induced acute kidney injury in a rat model of sepsis. Int. Urol. Nephrol. 2015, 47, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Lu, G. Thioredoxin relieves lipopolysaccharide-induced acute kidney injury in mice by reducing inflammation, oxidative stress and apoptosis. Exp. Ther. Med. 2021, 21, 629. [Google Scholar] [CrossRef]
- Soni, S.; Martens, M.D.; Takahara, S.; Silver, H.L.; Maayah, Z.H.; Ussher, J.R.; Ferdaoussi, M.; Dyck, J.R.B. Exogenous ketone ester administration attenuates systemic inflammation and reduces organ damage in a lipopolysaccharide model of sepsis. Biochim. Biophys. Acta Mol. Basis. Dis. 2022, 1868, 166507. [Google Scholar] [CrossRef] [PubMed]
- Weckx, R.; Goossens, C.; Derde, S.; Pauwels, L.; Vander Perre, S.; Van den Bergh, G.; Langouche, L. Identification of the toxic threshold of 3-hydroxybutyrate-sodium supplementation in septic mice. BMC Pharmacol. Toxicol. 2021, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Onur, B.; Cavusoglu, K.; Yalcin, E.; Acar, A. Paraquat toxicity in different cell types of swiss albino mice. Sci. Rep. 2022, 12, 4818. [Google Scholar] [CrossRef]
- Safaei Asl, A.; Dadashzadeh, P. Acute kidney injury in patients with paraquat intoxication; a case report and review of the literature. J. Renal. Inj. Prev. 2016, 5, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Fussell, K.C.; Udasin, R.G.; Gray, J.P.; Mishin, V.; Smith, P.J.; Heck, D.E.; Laskin, J.D. Redox cycling and increased oxygen utilization contribute to diquat-induced oxidative stress and cytotoxicity in chinese hamster ovary cells overexpressing nadph-cytochrome p450 reductase. Free Radic. Biol. Med. 2011, 50, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.J.; Lodge, J.K.; Traber, M.G.; Matsugo, S.; Packer, L. Comparison between copper-mediated and hypochlorite-mediated modifications of human low density lipoproteins evaluated by protein carbonyl formation. J. Lipid Res. 1997, 38, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Lodge, J.K.; Traber, M.G.; Packer, L. Apolipoprotein b carbonyl formation is enhanced by lipid peroxidation during copper-mediated oxidation of human low-density lipoproteins. Arch. Biochem. Biophys. 1997, 339, 165–171. [Google Scholar] [CrossRef]
- Wu, J.; Li, R.; Li, W.; Ren, M.; Thangthaeng, N.; Sumien, N.; Liu, R.; Yang, S.; Simpkins, J.W.; Forster, M.J.; et al. Administration of 5-methoxyindole-2-carboxylic acid that potentially targets mitochondrial dihydrolipoamide dehydrogenase confers cerebral preconditioning against ischemic stroke injury. Free Radic. Biol. Med. 2017, 113, 244–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Jin, Z.; Yang, X.; Yan, L.J. Post-ischemic administration of 5-methoxyindole-2-carboxylic acid at the onset of reperfusion affords neuroprotection against stroke injury by preserving mitochondrial function and attenuating oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 444–450. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Wang, Y.; Zhao, M. Nlrp3 inflammasome activation regulated by nf-kappab and dapk contributed to paraquat-induced acute kidney injury. Immunol. Res. 2017, 65, 687–698. [Google Scholar] [CrossRef]
- Iskander, A.; Yan, L.J. Cisplatin-induced kidney toxicity: Potential roles of major nad(+)-dependent enzymes and plant-derived natural products. Biomolecules 2022, 12, 1078. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Sears, S.M.; Orwick, A.; Siskind, L.J. Modeling cisplatin-induced kidney injury to increase translational potential. Nephron 2022, 147, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.N.; Yan, D.Q.; Zhang, X.P.; Chen, X.; Zhang, W.H.; Jia, H.L. Kidney mesenchymal stem cells alleviate cisplatin-induced kidney injury and apoptosis in rats. Tissue Cell 2023, 80, 101998. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Yang, M.; Han, Y.; Zhao, H.; Jiang, N.; Li, L.; Chen, W.; Li, C.; Yang, J.; Liu, Y.; et al. Beta-hydroxybutyrate against cisplatin-induced acute kidney injury via inhibiting nlrp3 inflammasome and oxidative stress. Int. Immunopharmacol. 2022, 111, 109101. [Google Scholar] [CrossRef]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Renal. Inj. Prev. 2015, 4, 20–27. [Google Scholar]
- Yan, Y.; Bai, J.; Zhou, X.; Tang, J.; Jiang, C.; Tolbert, E.; Bayliss, G.; Gong, R.; Zhao, T.C.; Zhuang, S. P2x7 receptor inhibition protects against ischemic acute kidney injury in mice. Am. J. Physiol. Cell Physiol. 2015, 308, C463–C472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Min, S.J.; Xu, B.; Zhang, C.; Pei, J.; Zhang, W.; Luo, G.H. The protective effects of exogenous spermine on renal ischemia-reperfusion injury in rats. Transl. Androl. Urol. 2021, 10, 2051–2066. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Targeting inflammation and oxidative stress as a therapy for ischemic kidney injury. Biochemistry 2020, 85, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jang, H.R. Role of t cells in ischemic acute kidney injury and repair. Korean J. Intern. Med. 2022, 37, 534–550. [Google Scholar] [CrossRef]
- Makievskaya, C.I.; Popkov, V.A.; Andrianova, N.V.; Liao, X.; Zorov, D.B.; Plotnikov, E.Y. Ketogenic diet and ketone bodies against ischemic injury: Targets, mechanisms, and therapeutic potential. Int. J. Mol. Sci. 2023, 24, 2576. [Google Scholar] [CrossRef]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameer, O.Z. Hypertension in chronic kidney disease: What lies behind the scene. Front. Pharmacol. 2022, 13, 949260. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Srinivas, T.; Ejaz, A.; Mu, W.; Roncal, C.; Sanchez-Lozada, L.G.; Gersch, M.; Rodriguez-Iturbe, B.; Kang, D.H.; et al. Essential hypertension, progressive renal disease, and uric acid: A pathogenetic link? J. Am. Soc. Nephrol. 2005, 16, 1909–1919. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.; McClure, T.S.; Koutnik, A.P.; Egan, B. Exogenous ketone supplements in athletic contexts: Past, present, and future. Sports Med. 2022, 52, 25–67. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Schnellmann, R.G. Pharmacological targeting of mitochondria in diabetic kidney disease. Pharmacol. Rev. 2023, 75, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Natesan, V.; Kim, S.J. Diabetic nephropathy-a review of risk factors, progression, mechanism, and dietary management. Biomol. Ther. 2021, 29, 365–372. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupsa, B.C.; Kibbey, R.G.; Inzucchi, S.E. Ketones: The double-edged sword of sglt2 inhibitors? Diabetologia 2023, 66, 23–32. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Maleki, M.; Butler, A.E.; Jamialahmadi, T.; Sahebkar, A. New insights into cellular links between sodium-glucose cotransporter-2 inhibitors and ketogenesis. J. Cell. Biochem. 2022, 123, 1879–1890. [Google Scholar] [CrossRef]
- Ito, M.; Tanaka, T. The anticipated renoprotective effects of sodium-glucose cotransporter 2 inhibitors. Intern. Med. 2018, 57, 2105–2114. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Aguero, J.; Tilemann, L.; Ladage, D.; Hammoudi, N.; Kawase, Y.; Santos-Gallego, C.G.; Fish, K.; Levine, R.A.; Hajjar, R.J. Characterizing preclinical models of ischemic heart failure: Differences between lad and lcx infarctions. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1478–H1486. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Mayr, M.; Badimon, J. Sglt2 inhibitors in heart failure: Targeted metabolomics and energetic metabolism. Circulation 2022, 146, 819–821. [Google Scholar] [CrossRef]
- Larmour, K.; Levin, A. Slowing progression in ckd: Dapa ckd and beyond. Clin. J. Am. Soc. Nephrol. 2021, 16, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.J.; Jardine, M.J.; Chertow, G.M.; Mahaffey, K.W. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: Lessons from credence and expectations from dapa-hf, dapa-ckd, and empa-kidney. Diabetes. Obes. Metab. 2020, 22 (Suppl. S1), 46–54. [Google Scholar] [CrossRef]
- Requena-Ibanez, J.A.; Santos-Gallego, C.G.; Rodriguez-Cordero, A.; Vargas-Delgado, A.P.; Badimon, J.J. Empagliflozin improves quality of life in nondiabetic hfref patients. Sub-analysis of the empatropism trial. Diabetes Metab. Syndr. 2022, 16, 102417. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Van Spall, H.G.C. In hfref, adding empagliflozin to medical therapy reduced a composite outcome, regardless of ckd status. Ann. Intern. Med. 2021, 174, JC68. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Chen, B.; Gong, A.Y.; Malhotra, D.K.; Gupta, R.; Dworkin, L.D.; Gong, R. The ketone body beta-hydroxybutyrate mitigates the senescence response of glomerular podocytes to diabetic insults. Kidney Int. 2021, 100, 1037–1053. [Google Scholar] [CrossRef]
- Kume, S. Ketone bodies: Back to a place in the sun. Kidney Int. 2021, 100, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.N.; Moon, J.H.; Cho, Y.M. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced nrf2 activation. Diabetes Obes. Metab. 2021, 23, 2561–2571. [Google Scholar] [CrossRef]
- Uddin, M.J.; Farjana, M.; Moni, A.; Hossain, K.S.; Hannan, M.A.; Ha, H. Prospective pharmacological potential of resveratrol in delaying kidney aging. Int. J. Mol. Sci. 2021, 22, 8258. [Google Scholar] [CrossRef] [PubMed]
- Chang-Panesso, M. Acute kidney injury and aging. Pediatr. Nephrol. 2021, 36, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yuan, L.; Liu, F.; Li, L.; Liu, J.; Chen, Y.; Lu, Y.; Yuan, Y. Molecular mechanisms and physiological functions of autophagy in kidney diseases. Front. Pharmacol. 2022, 13, 974829. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Tian, Z.; Ma, K.; Li, Y.; Wang, L.; Nasser, M.I.; Liu, C. The diseased kidney: Aging and senescent immunology. Immun. Ageing 2022, 19, 58. [Google Scholar] [CrossRef]
- Guo, J.; Zheng, H.J.; Zhang, W.; Lou, W.; Xia, C.; Han, X.T.; Huang, W.J.; Zhang, F.; Wang, Y.; Liu, W.J. Accelerated kidney aging in diabetes mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 1234059. [Google Scholar] [CrossRef]
- Sohal, R.S.; Forster, M.J. Caloric restriction and the aging process: A critique. Free Radic. Biol. Med. 2014, 73, 366–382. [Google Scholar] [CrossRef] [Green Version]
- Dubey, A.; Forster, M.J.; Lal, H.; Sohal, R.S. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch. Biochem. Biophys. 1996, 333, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Rebrin, I.; Forster, M.J.; Sohal, R.S. Effects of age and caloric restriction on mitochondrial protein oxidative damage in mice. Mech. Ageing Dev. 2012, 133, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mank, M.M.; Reed, L.F.; Walton, C.J.; Barup, M.L.T.; Ather, J.L.; Poynter, M.E. Therapeutic ketosis decreases methacholine hyperresponsiveness in mouse models of inherent obese asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L243–L257. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Cunningham, R.P.; Davis, R.A.H.; Deemer, S.E.; Roberts, B.M.; Plaisance, E.P.; Rector, R.S. A dietary ketone ester mitigates histological outcomes of nafld and markers of fibrosis in high-fat diet fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G564–G572. [Google Scholar] [CrossRef] [PubMed]
- Izuta, Y.; Imada, T.; Hisamura, R.; Oonishi, E.; Nakamura, S.; Inagaki, E.; Ito, M.; Soga, T.; Tsubota, K. Ketone body 3-hydroxybutyrate mimics calorie restriction via the nrf2 activator, fumarate, in the retina. Aging Cell 2018, 17, e12699. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Xu, G.; Zhu, Y.; Lin, C.; Yan, Z.; Shao, S. Association of body mass index and acute kidney injury incidence and outcome: A systematic review and meta-analysis. J. Ren. Nutr. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Bao, Y.W.; Yuan, Y.; Chen, J.H.; Lin, W.Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool Res. 2018, 39, 72–86. [Google Scholar]
- Pillai, A.; Fulmali, D. A narrative review of new treatment options for diabetic nephropathy. Cureus 2023, 15, e33235. [Google Scholar] [CrossRef]
- Georgianos, P.I.; Vaios, V.; Eleftheriadis, T.; Papachristou, E.; Liakopoulos, V. Therapeutic advances in diabetic kidney disease. Int. J. Mol. Sci. 2023, 24, 2803. [Google Scholar] [CrossRef]
- Guo, M.; Chen, Q.; Huang, Y.; Wu, Q.; Zeng, Y.; Tan, X.; Teng, F.; Ma, X.; Pu, Y.; Huang, W.; et al. High glucose-induced kidney injury via activation of necroptosis in diabetic kidney disease. Oxid. Med. Cell. Longev. 2023, 2023, 2713864. [Google Scholar] [CrossRef]
- Kelsey, R. Metabolic changes in the kidney. Nat. Rev. Nephrol. 2020, 16, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fang, X.; Zhang, H.; Gao, W.; Hsu, H.J.; Roman, R.J.; Fan, F. Genetic susceptibility of hypertension-induced kidney disease. Physiol. Rep. 2021, 9, e14688. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Bajo, M.A.; Selgas, R.; Diez, J.J. Thyroid dysfunction and kidney disease: An update. Rev. Endocr. Metab. Disord. 2017, 18, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Sazgar, M. Kidney disease and epilepsy. J. Stroke Cerebrovasc. Dis. 2021, 30, 105651. [Google Scholar] [CrossRef]
- Roy, P.J.; Weltman, M.; Dember, L.M.; Liebschutz, J.; Jhamb, M.; Consortium, H. Pain management in patients with chronic kidney disease and end-stage kidney disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 671–680. [Google Scholar] [CrossRef]
- Langston, C. Managing fluid and electrolyte disorders in kidney disease. Vet. Clin. N. Am. Small Anim. Pract. 2017, 47, 471–490. [Google Scholar] [CrossRef]
- Collister, D.; Rigatto, C.; Tangri, N. Anemia management in chronic kidney disease and dialysis: A narrative review. Curr. Opin. Nephrol. Hypertens. 2017, 26, 214–218. [Google Scholar] [CrossRef]
- Woo, Y.M.; Pereira, B.J.; Gill, J.S. Chronic kidney disease progression in native and transplant kidneys. Curr. Opin. Nephrol. Hypertens. 2004, 13, 607–611. [Google Scholar] [CrossRef]
- Strubl, S.; Oehm, S.; Torres, J.A.; Grundmann, F.; Haratani, J.; Decker, M.; Vuong, S.; Kaur Bhandal, A.; Methot, N.; Haynie-Cion, R.; et al. Ketogenic dietary interventions in autosomal dominant polycystic kidney disease-a retrospective case series study: First insights into feasibility, safety and effects. Clin. Kidney J. 2022, 15, 1079–1092. [Google Scholar] [CrossRef]
- Kosinski, C.; Jornayvaz, F.R. Effects of ketogenic diets on cardiovascular risk factors: Evidence from animal and human studies. Nutrients 2017, 9, 517. [Google Scholar] [CrossRef] [Green Version]
- Sampath, A.; Kossoff, E.H.; Furth, S.L.; Pyzik, P.L.; Vining, E.P. Kidney stones and the ketogenic diet: Risk factors and prevention. J. Child Neurol. 2007, 22, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Sumien, N.; Thangthaeng, N.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013, 47, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Jin, Z.; Yan, L.J. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox. Biol. 2017, 11, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dendooven, A.; Ishola, D.A., Jr.; Nguyen, T.Q.; Van der Giezen, D.M.; Kok, R.J.; Goldschmeding, R.; Joles, J.A. Oxidative stress in obstructive nephropathy. Int. J. Exp. Pathol. 2011, 92, 202–210. [Google Scholar] [CrossRef]
- Brosey, C.A.; Ho, C.; Long, W.Z.; Singh, S.; Burnett, K.; Hura, G.L.; Nix, J.C.; Bowman, G.R.; Ellenberger, T.; Tainer, J.A. Defining nadh-driven allostery regulating apoptosis-inducing factor. Structure 2016, 24, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Zhang, Y.; Xiong, Y.; Zhu, Z.; Wang, L.; Liu, X. Mitochondria-targeted antioxidant mitoquinone maintains mitochondrial homeostasis through the sirt3-dependent pathway to mitigate oxidative damage caused by renal ischemia/reperfusion. Oxid. Med. Cell. Longev. 2022, 2022, 2213503. [Google Scholar] [CrossRef]
- Qin, L.; Xi, S. The role of mitochondrial fission proteins in mitochondrial dynamics in kidney disease. Int. J. Mol. Sci. 2022, 23, 14725. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Yan, L.-J. The Role of Ketone Bodies in Various Animal Models of Kidney Disease. Endocrines 2023, 4, 236-249. https://doi.org/10.3390/endocrines4010019
Liu H, Yan L-J. The Role of Ketone Bodies in Various Animal Models of Kidney Disease. Endocrines. 2023; 4(1):236-249. https://doi.org/10.3390/endocrines4010019
Chicago/Turabian StyleLiu, Haoxin, and Liang-Jun Yan. 2023. "The Role of Ketone Bodies in Various Animal Models of Kidney Disease" Endocrines 4, no. 1: 236-249. https://doi.org/10.3390/endocrines4010019
APA StyleLiu, H., & Yan, L.-J. (2023). The Role of Ketone Bodies in Various Animal Models of Kidney Disease. Endocrines, 4(1), 236-249. https://doi.org/10.3390/endocrines4010019