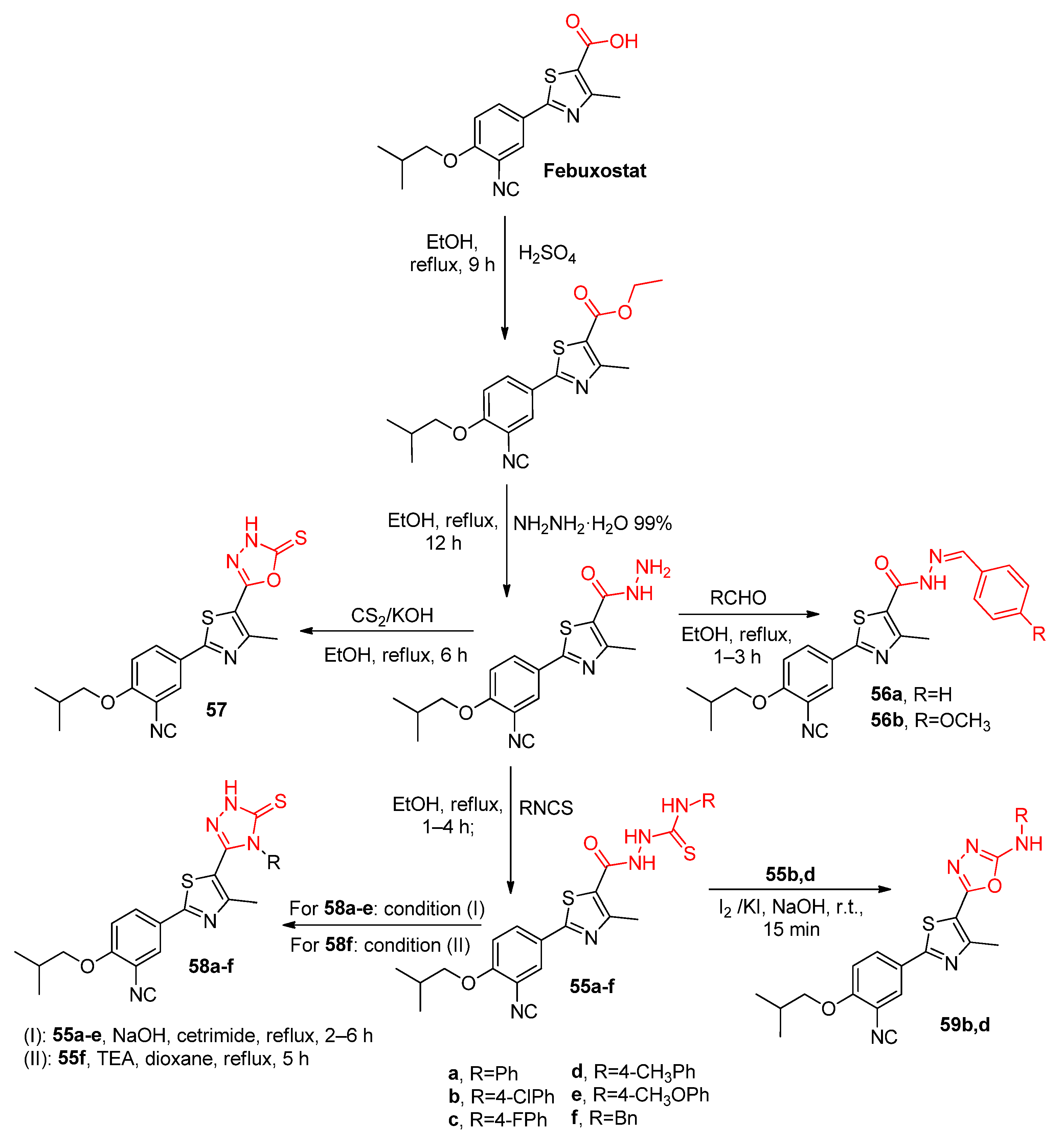
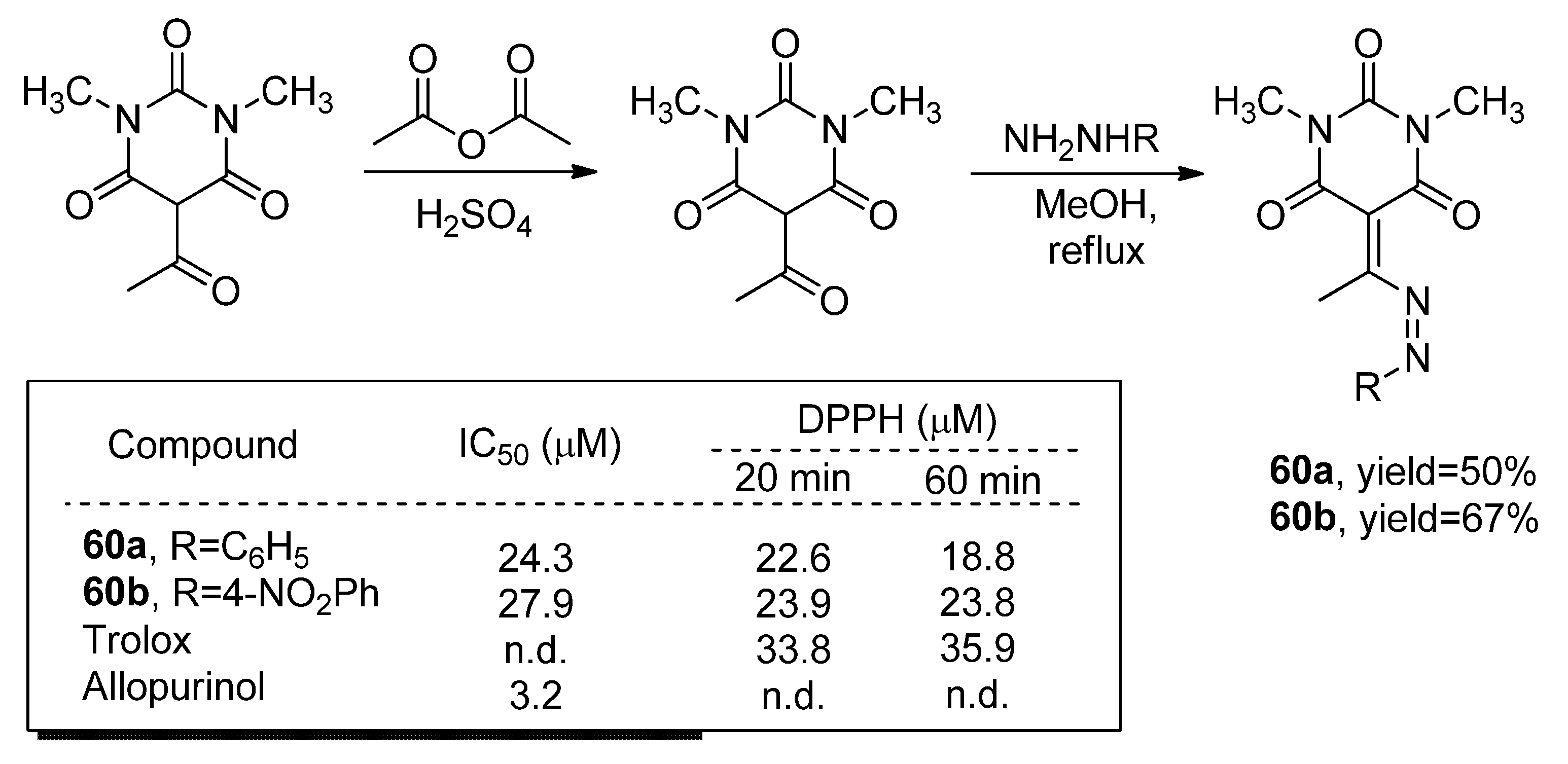
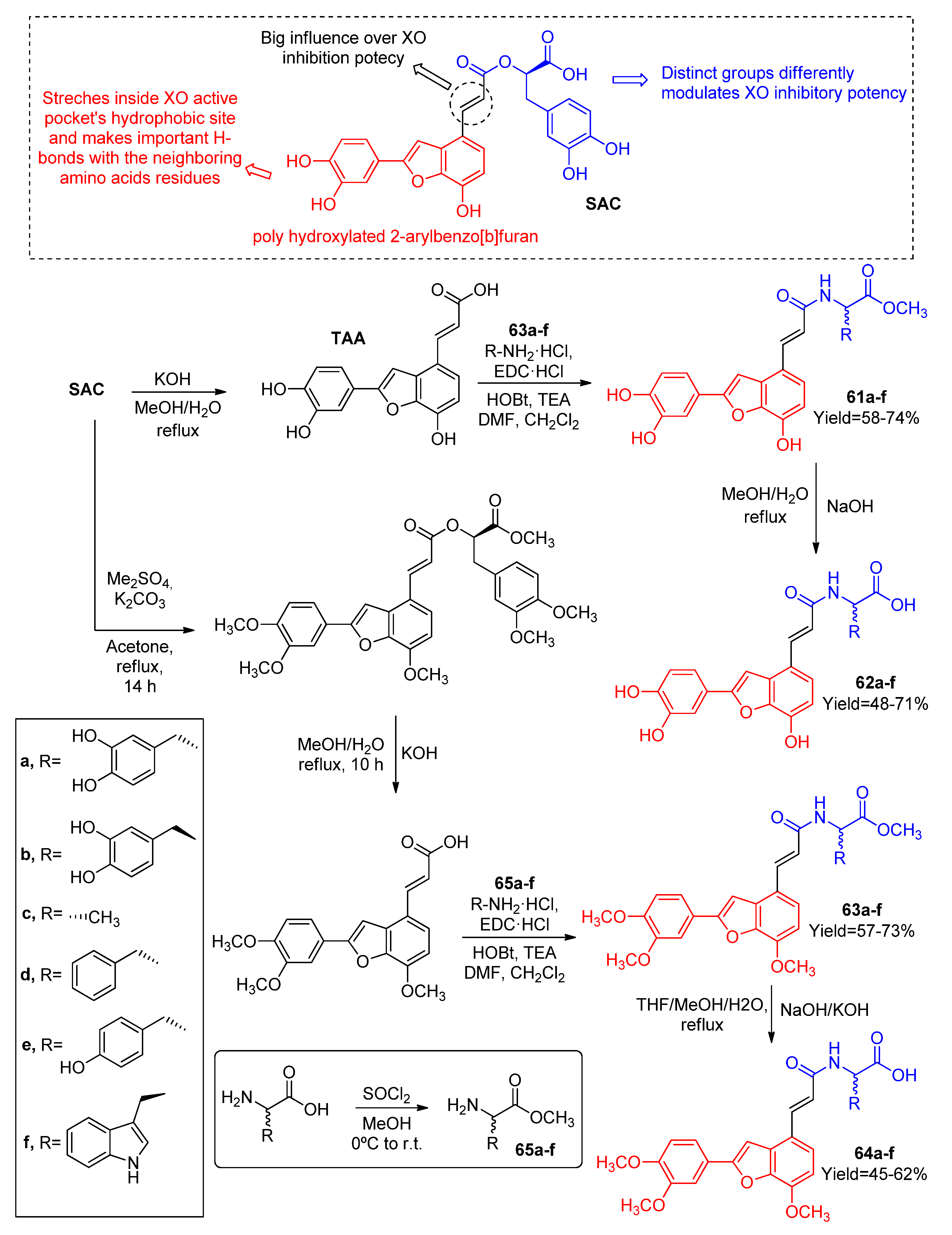
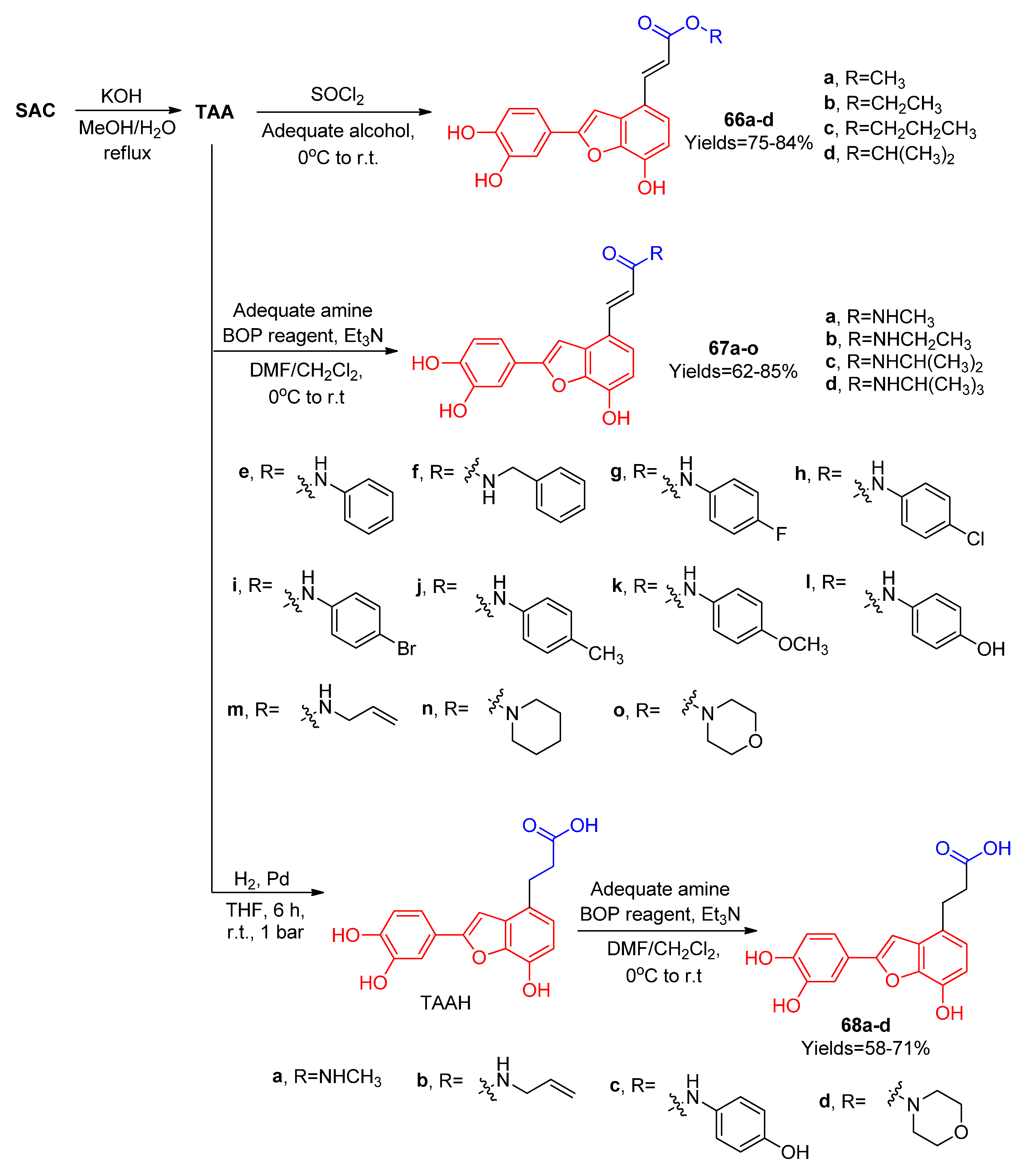
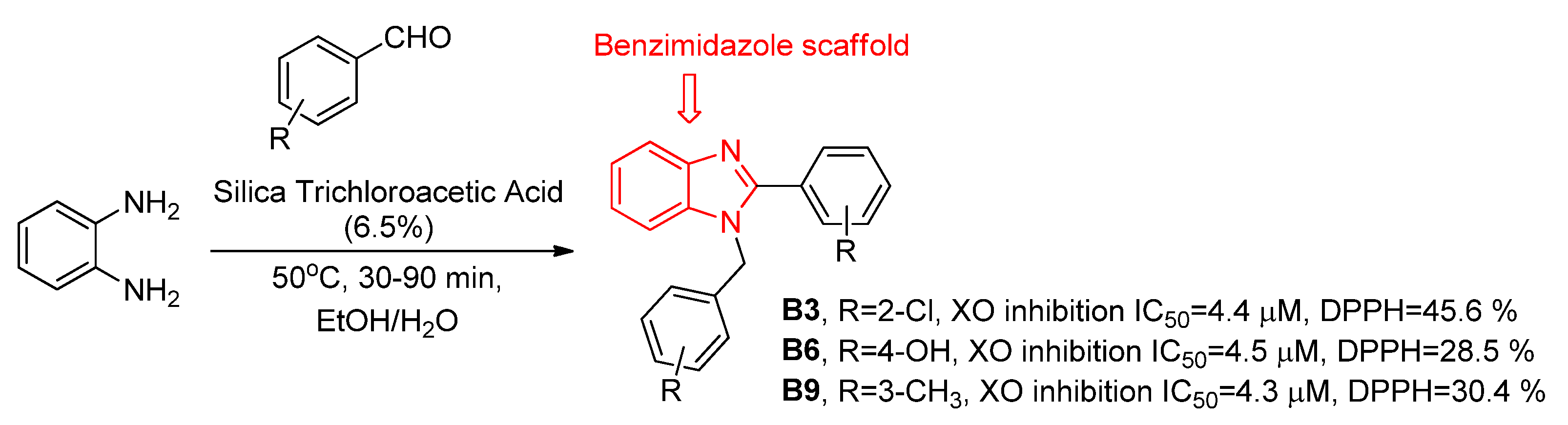
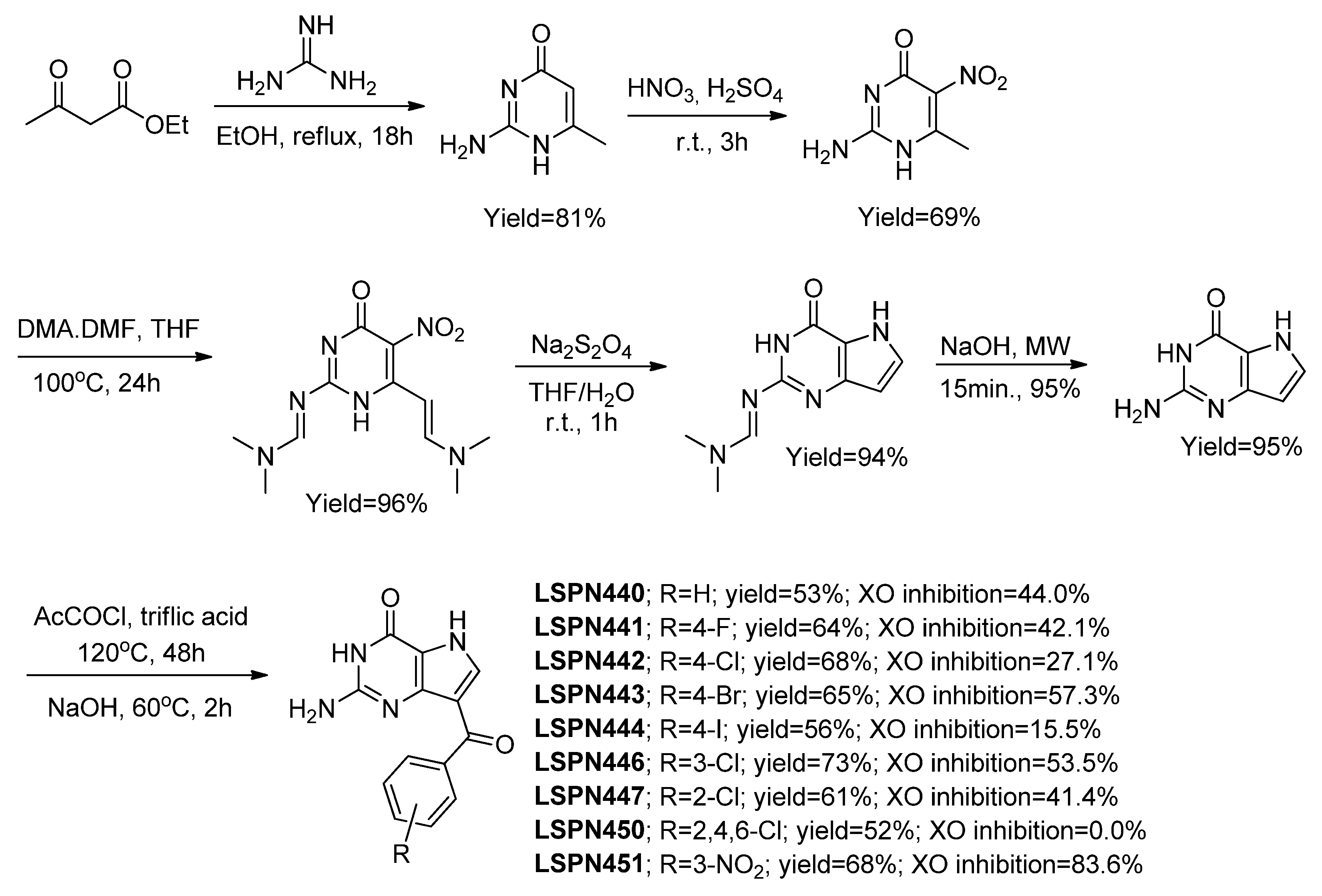
3.1.1. Derivatives Based on Five-Member Heterocyclic Rings
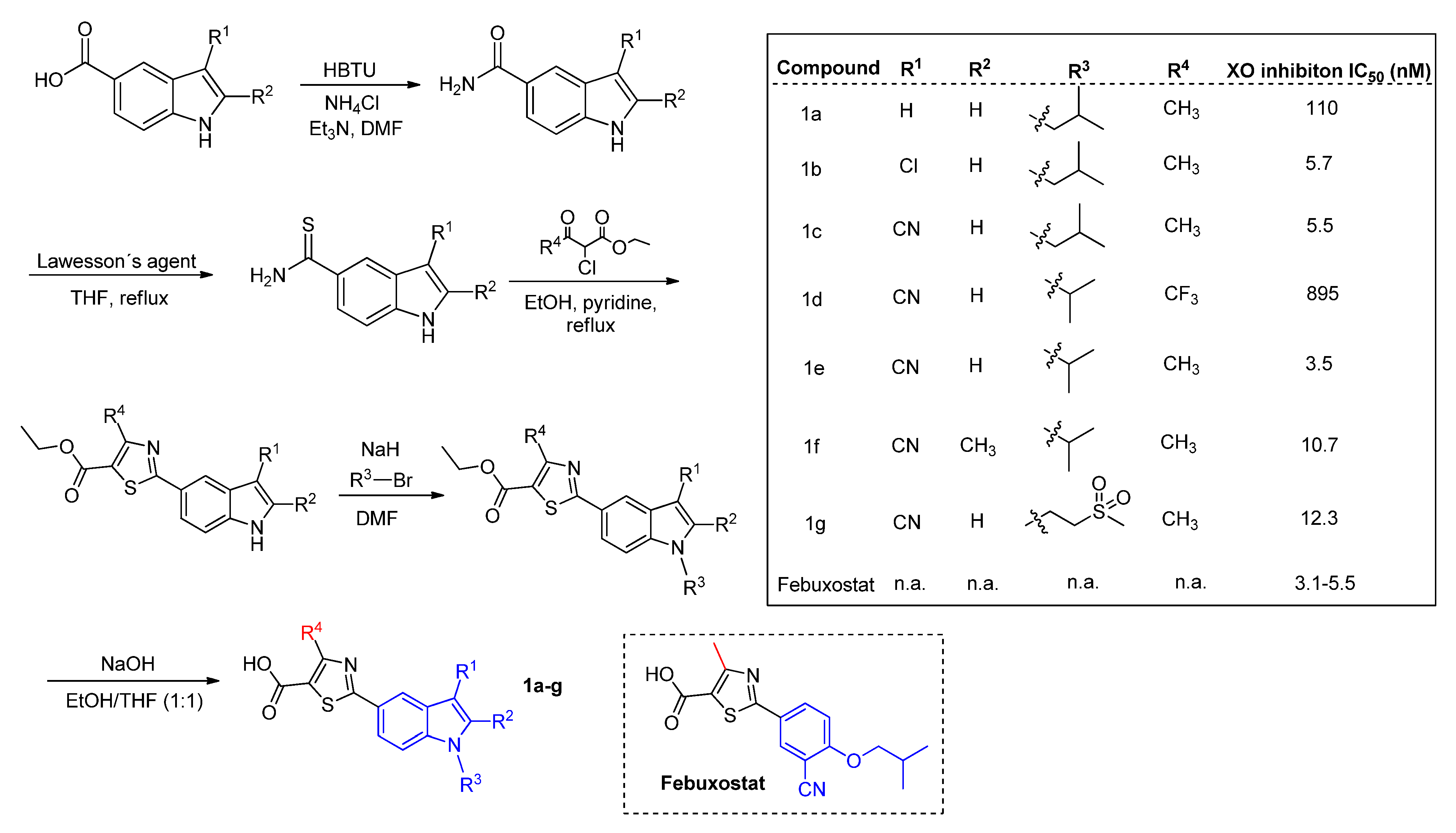
Focusing on a structure activity relationship (SAR) analysis, a library of more than twenty hybrid structures of indolethiazoles with four different substituents were explored as XO inhibitors and urate-lowering activity in rats by Song and coworkers, of which we highlight the selected examples
1a–
g (
Scheme 2) [
38]. Firstly, it was shown that electron-withdrawing substituents at the R
1-position favor XO inhibition. While at the R
4-position, it harshly decreases. The activity of the molecule is also favored with hydrophobic substituents at the R
3-position and without steric hindrance between R
2-R
3-positions. The best compound tested (
1e) also showed a uric acid inhibition of 60% at 1 h (10 mg/kg) in rats, beyond the excellent in vitro IC
50 of 3.5 nM, comparable to febuxostat. The other derivatives also reached nanomolar IC
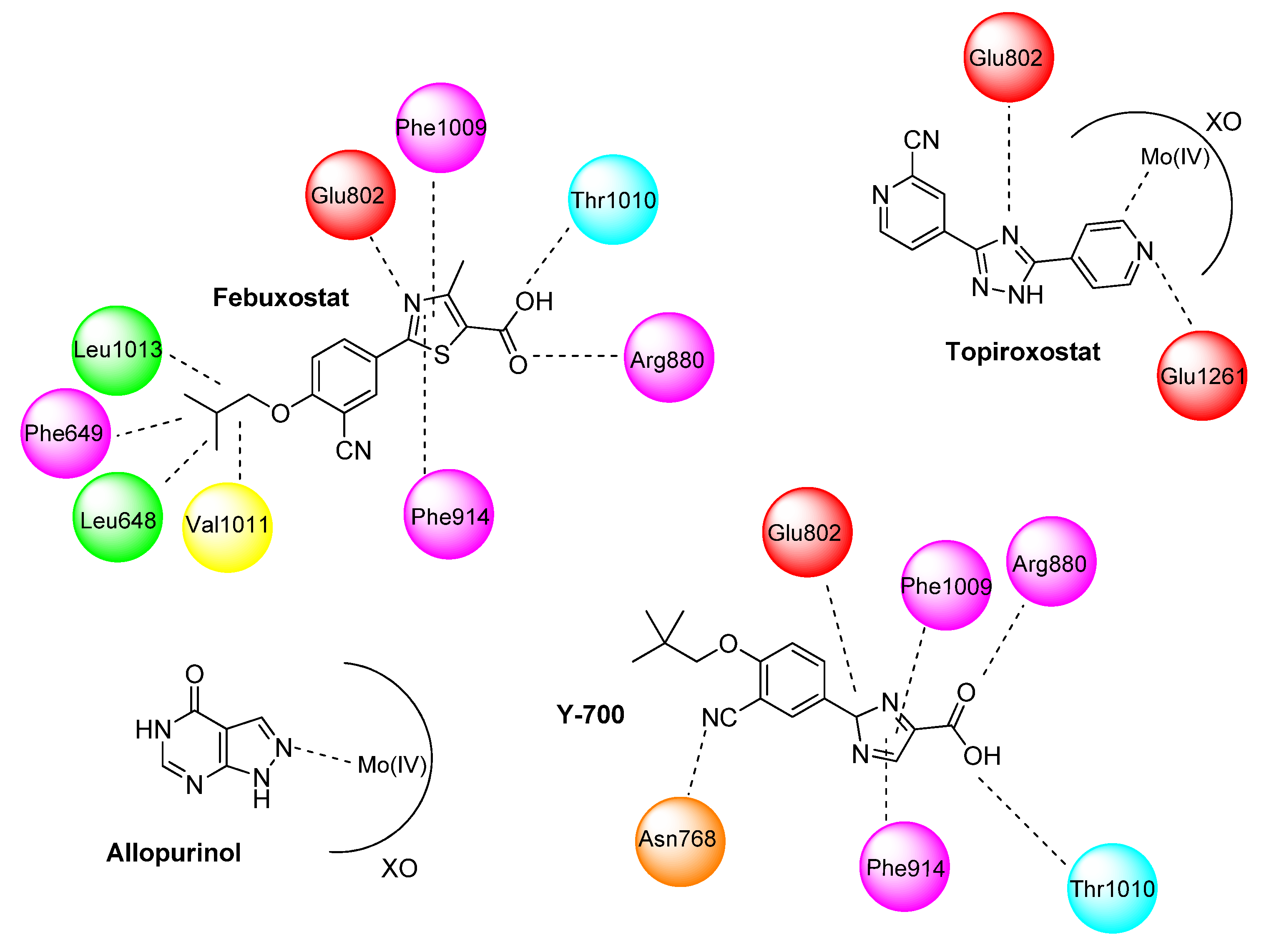
50 values. A docking analysis shows the same interactions of the carboxylate moiety and the thiazole ring that occurs with febuxostat. Despite the replacement of the phenyl group by an indole group, the cyano moiety was able to form hydrogen bonds with an Asn768 residue.
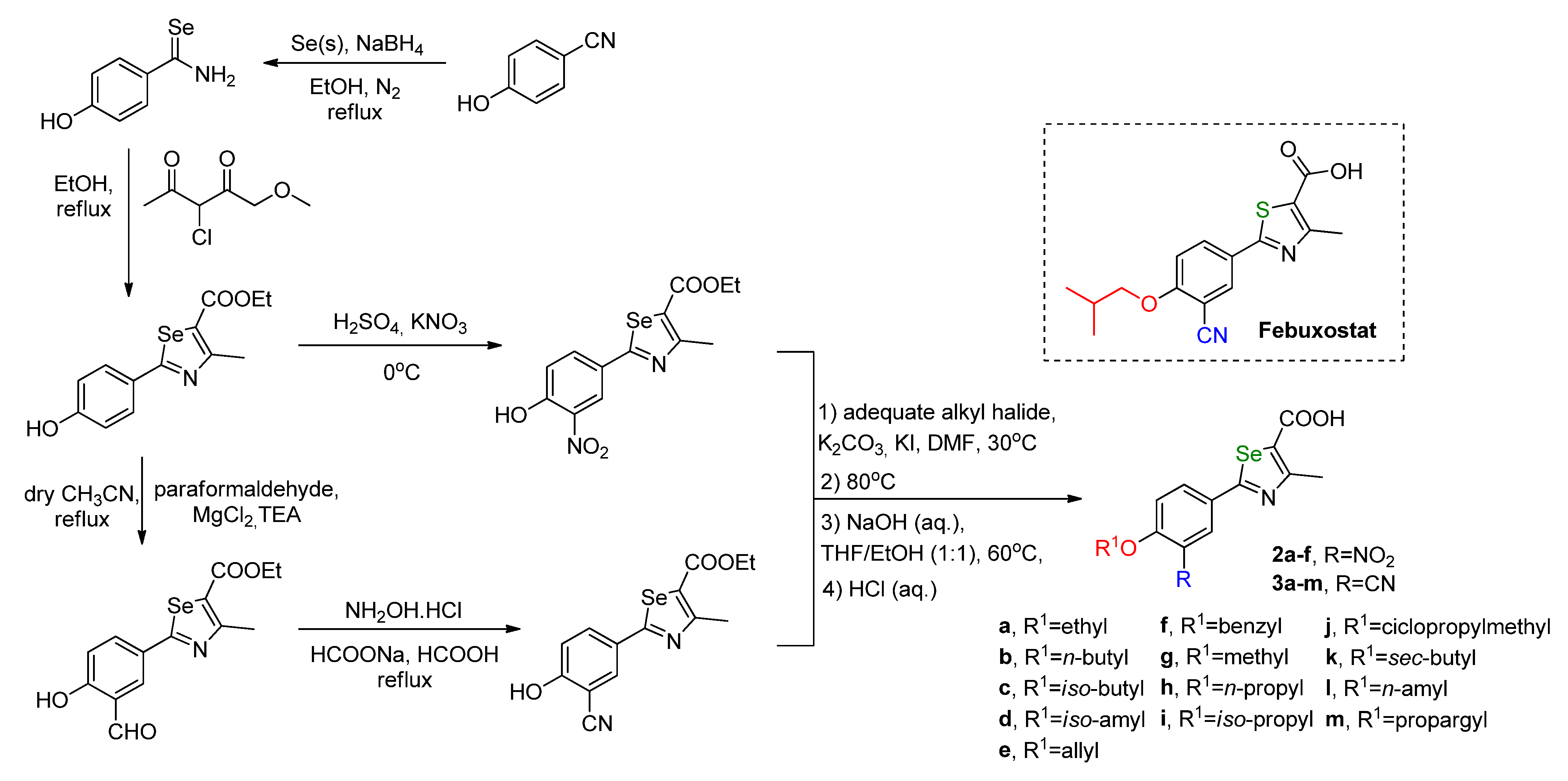
The replacement of the sulfur atom by a selenium one, in a series of febuxostat derivatives,
2a–
f and
3a–
m (
Scheme 3), revealed an improvement in the inhibition potential of XO with in vitro IC
50 values in nanomolar ranges (same as febuxostat), as observed by Guan and coworkers (
Table 1) [
39]. The most potent XO inhibitor (
3e) was compared with febuxostat (IC
50 of 18.6 nM), showing a better performance with an in vitro IC
50 value of 5.5 nM. Interestingly, its non-hydrolyzed precursor (not represented) did not present any activity at the tested concentration of 10 μM. Despite the size difference between sulfur and selenium atoms, a docking analysis showed similar interactions with the XO active site and a good overlap of the molecules. The kinetics analysis for
3e showed a mixed-type inhibition.
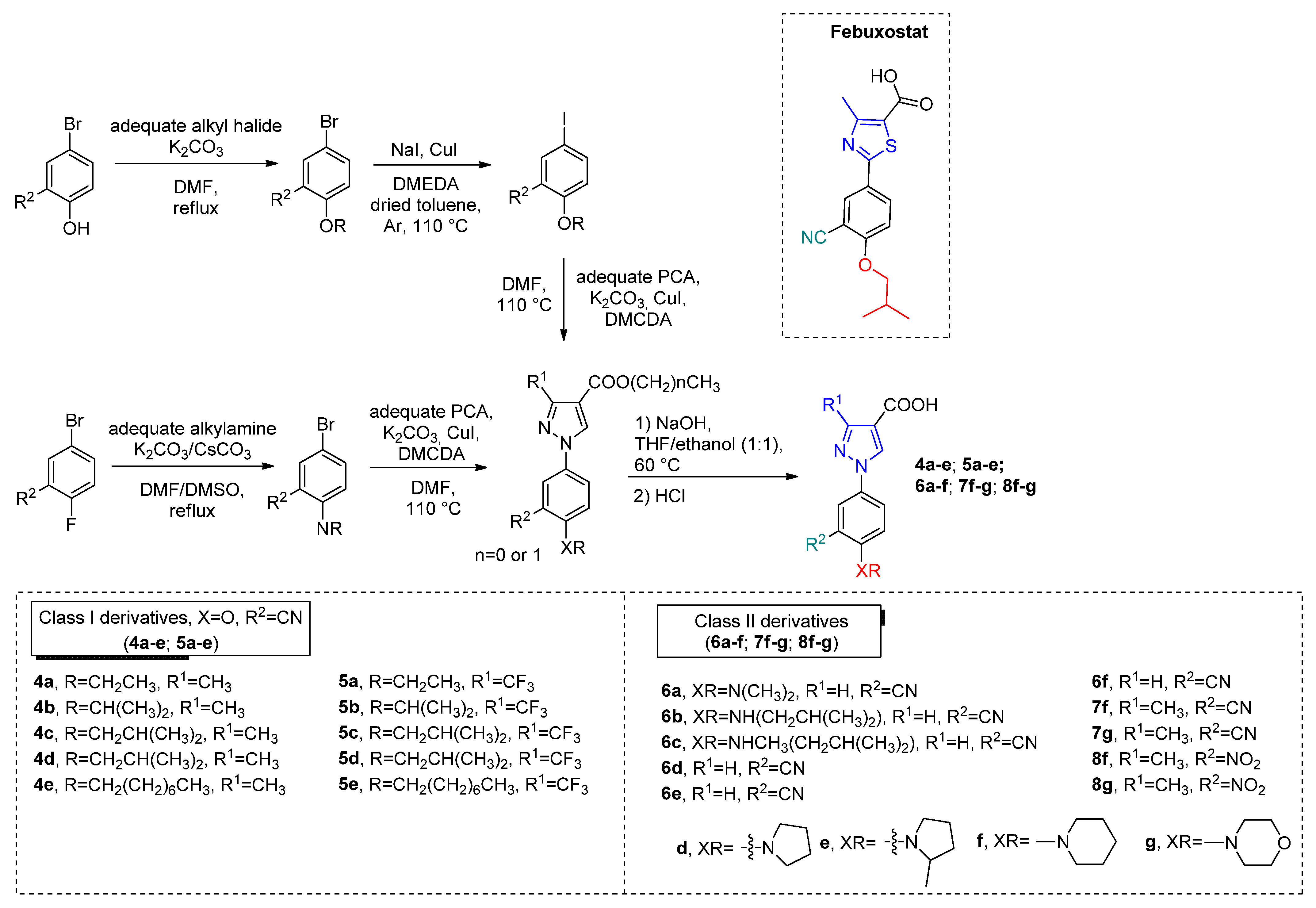
Other two classes (CI =
4a–
e,
5a–
e, and CII =
6a–
f,
7f,
g,
8f,
g) of Febuxostat analogs were synthesized by Jing Li and coworkers [
40] to explore the isosteric replacement of the thiazole ring with a pyrazole ring (
Scheme 4). Modifications between class I and II were mainly centered on phenyl substituents (-OR for CI and -NR for CII) for SAR studies. For CI class, substituents at R
1 proved to be inefficient for the inhibition effect, once the derivatives did not show significant activity for the tested concentration (
Table 2). While for CII, the general observed IC
50 values were comparable to febuxostat when R
1 = H. Moreover, for the most potent compound,
6f (IC
50 = 4.2 nM), molecular modeling showed similar interactions with the XO active site as well as Y-700 and febuxostat. Additionally, the pyrazole ring, as with the thiazole ring in febuxostat, exhibits aromatic interactions with Phe914 and Phe1009. Studies in vivo have shown that oral administration of
6f reduced serum uric acid levels in hyperuricemic mice with a similar profile of febuxostat.
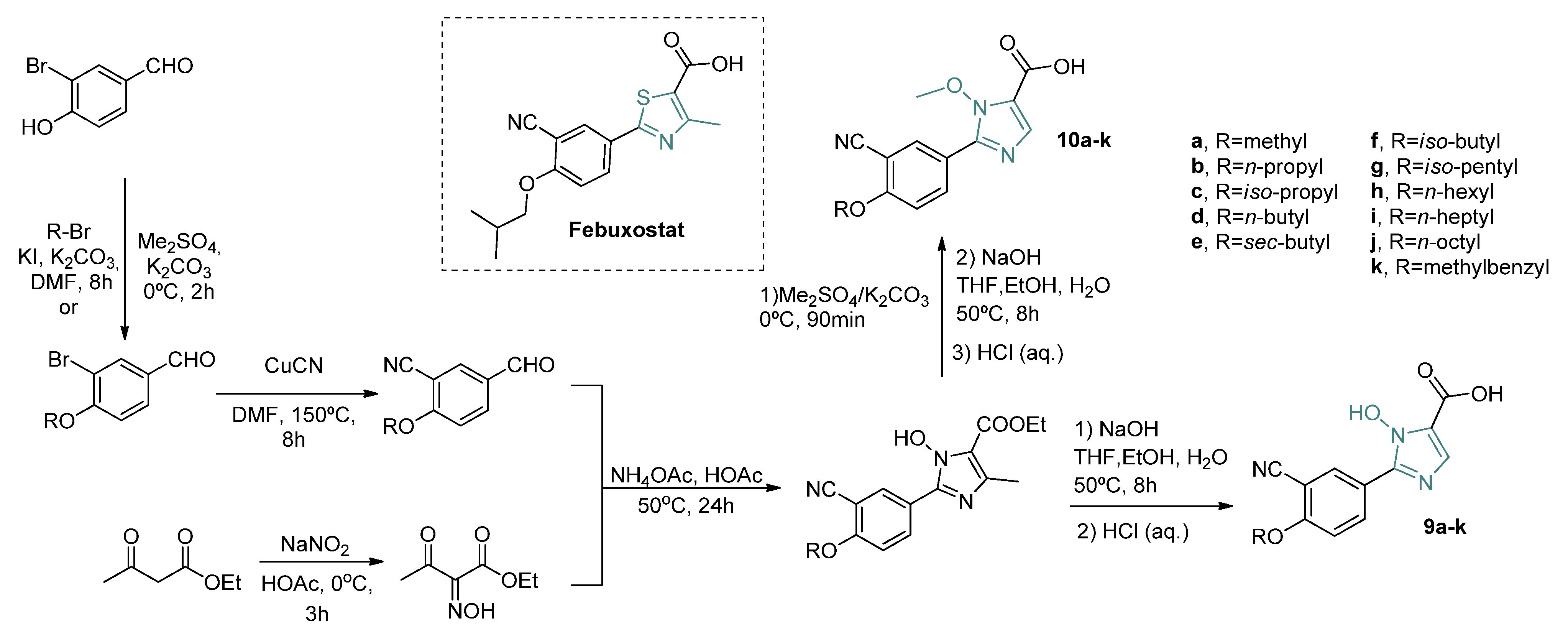
Another deviation in the five-member ring, proposed by Chen and coworkers [
41], comprised the replacement of the thiazole group for the 1-hydroxy/methoxy-imidazole ring (
Scheme 5). The in vitro IC
50 of XO inhibition of the hydroxy-imidazole (
9a–
k) class presented significatively lower values than the methoxy-imidazole class (
10a–
k) (
Table 3). Improvement of the activity was found for intermediary carbon chain length substituents at the 4-position (
n-butoxy, sec-butoxy, and
iso-butoxy). Docking analysis of one of the most potent compounds tested (
9f, in vitro IC
50 of 6 nM) showed that the hydroxyl group bonded to the nitrogen atom of the imidazole ring improved the interaction with the active site of XO through an additional hydrogen bond with Thr1010. Kinetic assays revealed that the compound
9f inhibition mechanism for XO was a mixed type.
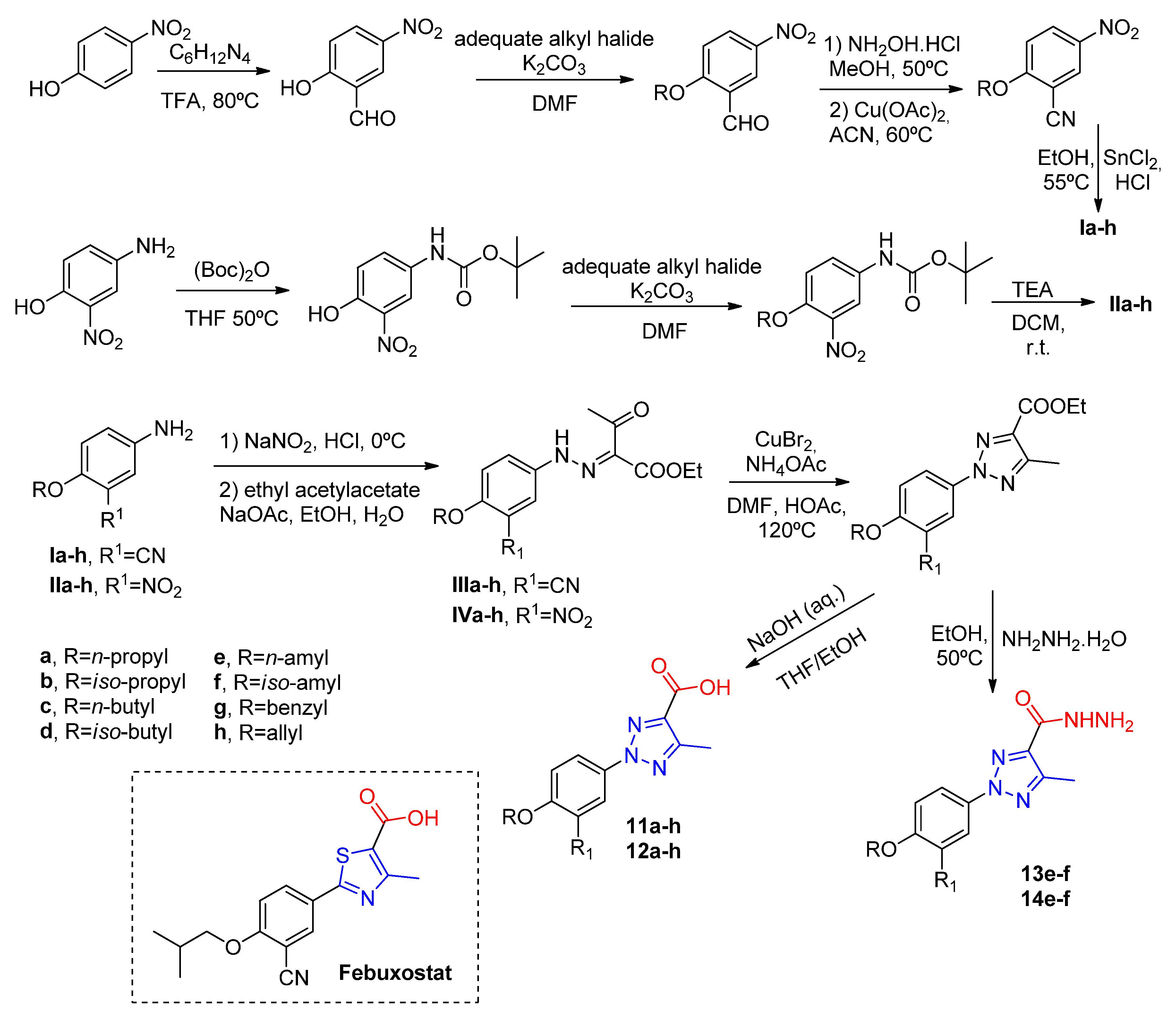
Following the same strategy, Shi and coworkers [
42] have synthesized two classes of compounds by replacing the thiazole ring of febuxostat with a 1,2,3-triazole ring containing a carboxylic acid (
11a–
h,
12a–
h) or a carbohydrazide substituent (
13e,
f,
14e,
f) (
Scheme 6). Among the tested compounds, those with carboxyl moiety presented in vitro IC
50 values ranging between 84–254 nM, while those compounds with carbohydrazide substituents were not active for XO inhibition (
Table 4). The most active compounds were those with
i-amyl at the R-position, and cyano group or nitro group at R
1 (
11f, in vitro IC
50 = 84 nM and
12f, in vitro IC
50 = 109 nM). However, when compared to febuxostat (in vitro IC
50 = 12 nM), those compounds displayed a slight decrease in inhibition potential. A docking analysis reveals similar interactions found in febuxostat and, in addition, it was observed that the nitrogen atom at the 3-position of triazole forms a hydrogen bond with Glu802 and an additional π–π interaction of the five-member ring with Phe1009.
Zhang and coworkers [
43] have also synthesized, tested, and compared febuxostat analogs
15a–
s containing the 1,2,3-triazole nucleus as XO inhibitors (
Scheme 7) to allopurinol, Y-700, and febuxostat as positive controls. However, in this case, the difference compared to the previously discussed Shi and coworkers’ work [
42] is in the isosteric replacement of the nitrogen atoms. The proximity of the X
1-position with molybdenum-pterin inspired the authors to replace a carbon atom at this position with a nitrogen atom. The majority of the tested compounds presented an in vitro IC
50 value lower than allopurinol (7.56 μM), but greater than Y-700 (0.016 μM) (
Table 5). SAR analysis showed that the lipophilicity at the R may be beneficial for the activity. The increase of the carbon chain from three to eight at the R leads to a significant increase in XO inhibition potential (IC
50 > 30 μM to IC
50 = 0.63 μM). An improvement of inhibition activity was also achieved by insertion of benzyl groups as R, as the
meta-Methoxybenzyl group (
15s) presented the lower in vitro IC
50 value of 0.21 μM. Docking analysis showed a similar interaction compared to febuxostat and the active site of XO, including the triazole ring aromatic interaction with Phe1009 and Phe914. However, the nitrogen atom at X
2-position does not seem to have any interaction with molybdenum-pterin as expected. Moreover, neighboring X
2-position nitrogen seemed to increase the hydrophilicity around the X
1-position nitrogen, leading to the observed decrease in derivatives potency.
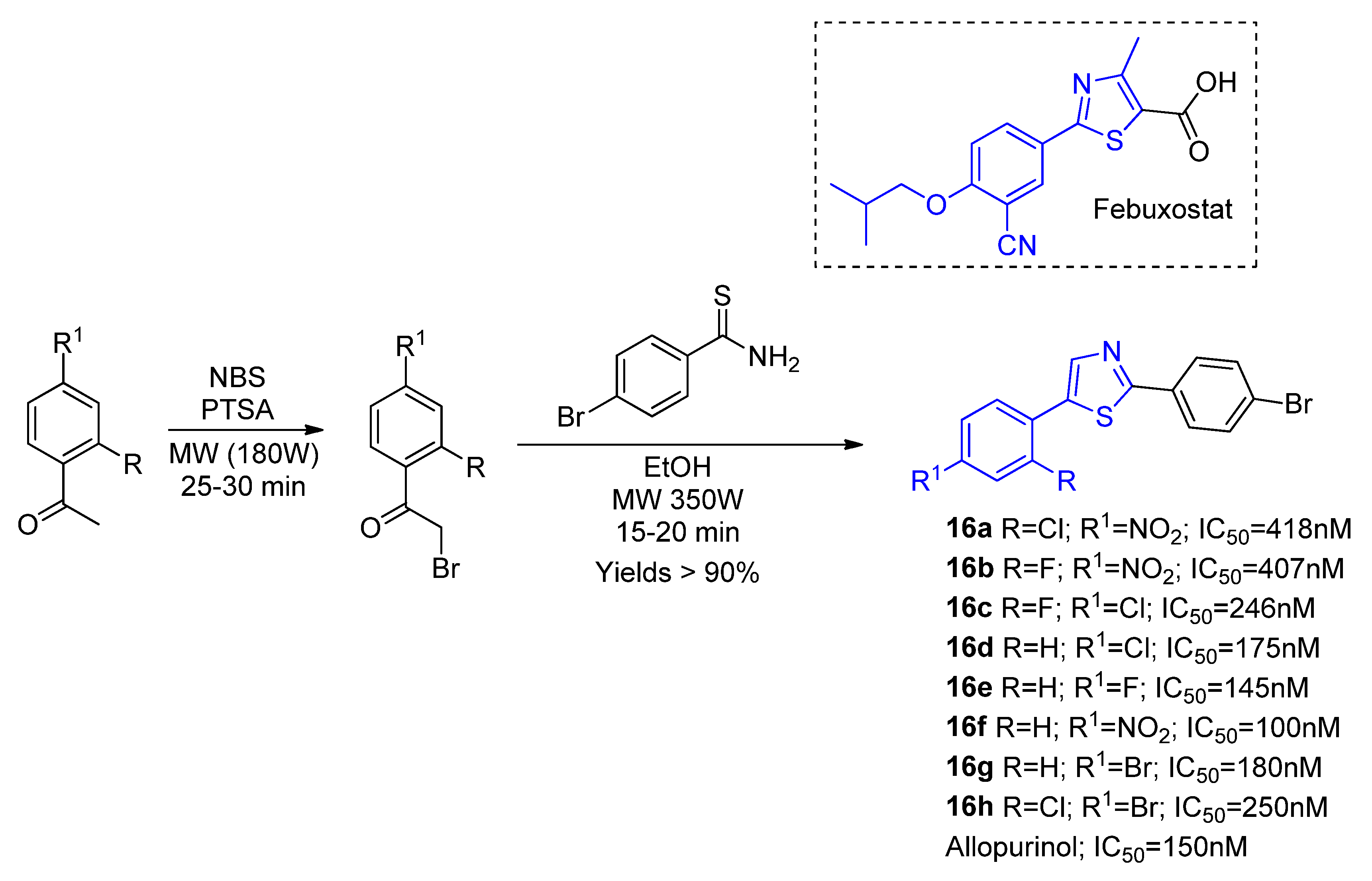
Exploring a microwave-assisted synthetic strategy, Jyothi and coworkers [
44] produced eight novel XO inhibitors (
16a–
h) comprising febuxostat’s thiazole nucleus through a highly efficient two-step procedure (
Scheme 8). The group had also tested the same reaction conditions under thermal heating, in which case the reactions needed between three to five hours and the yields dropped to 64–76%. All the synthesized compounds presented in vitro XO inhibition activities in the nanomolar range. However, the derivatives
16e,
f showed the greatest potency, with lower IC
50 values (IC
50 = 145 nm and 100 nM, respectively) than the control drug allopurinol (IC
50 = 150 nm). A SAR investigation pointed out that electron withdrawing groups at the phenyl ring
para position are beneficial for the inhibitory activity improvement. Molecular docking with the most active compound
16f pointed out a stronger binding affinity (−9.1 kcal/mol) compared to allopurinol (−7.0 kcal/mol) and more non-binding interaction with amino acids residues within the XO active site, which lead to a more stable bond with the enzyme. A molecular dynamic assay pointed out that there were no significative structural changes from the docked model.
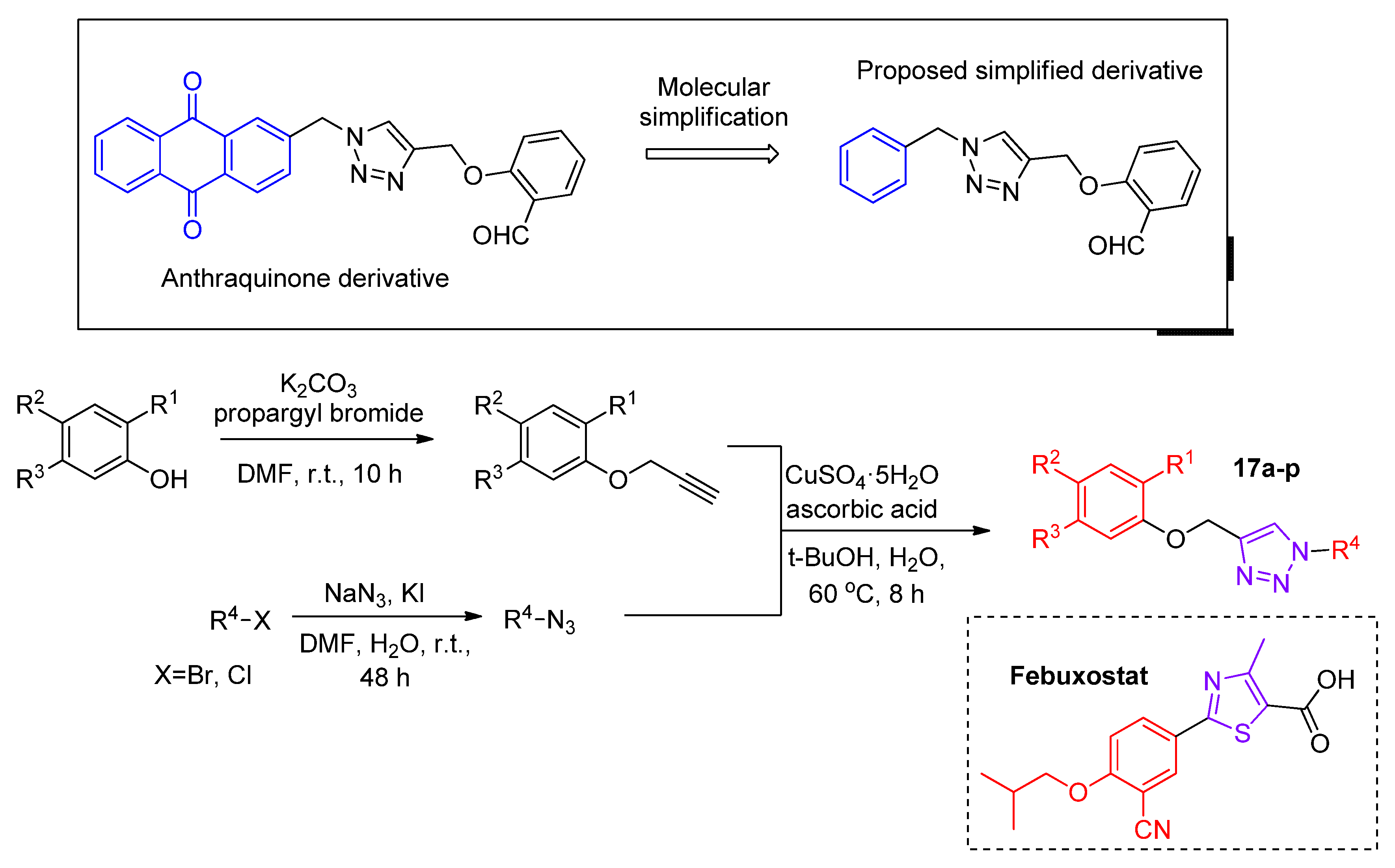
Based in the molecular skeleton of an anthraquinone derivative previously synthesized by the group, which presented a promising XO inhibition activity of 0.61 mM, Zhang and coworkers [
45] developed novel XO inhibitors. Two of the group’s major concerns regarding the anthraquinone based derivative were the high lipophilicity giving a poor drug-like characteristic and the DNA binding affinity leading to potential collateral cytotoxic effects. Thus, molecular deviations were proposed, and more than thirty-five compounds were obtained. We present the most active ones and some analogues
17a–
p to discuss important SAR characteristics (
Scheme 9). The best IC
50 values were achieved with either the introduction of a fluorine atom at the benzaldehyde R
2 position and/or a
meta-methoxybenzyl at R
4 position (
Table 6). The absence of aldehyde functionalization at R
1 position resulted in inactive compounds for the chosen criteria of 25 μM maximum concentration for the in vitro activity assays. Moreover, docking analysis showed hydrophobic interactions of the
meta-methoxybenzyl with Phe649 and Phe1013 and a well-adjusted accommodation of the
meta-fluorine benzaldehyde moiety at the hydrophobic pocket of XO composed of Leu873, Ala1078, and Val1011 amino acids, justifying the promising results comprising those deviations. The parameters metrics of ligand efficiency (LE) and lipophilic ligand efficiency (LLE) were employed to correlate biological activity with the lipophilicity of the most potent compounds showing an important improvement of those parameters with respect to allopurinol used as a positive control, including high solubility in the buffer. Additionally, in vivo tests with the most potent compound
17h showed a 31% of serum uric acid levels reduction in vehicle-treated rats at a dose of 20 mg/kg after 2 h compared to the control, which highlights the selected compound as a promising lead compound.
3.1.2. Derivatives Based on Six-Member Heterocyclic Rings
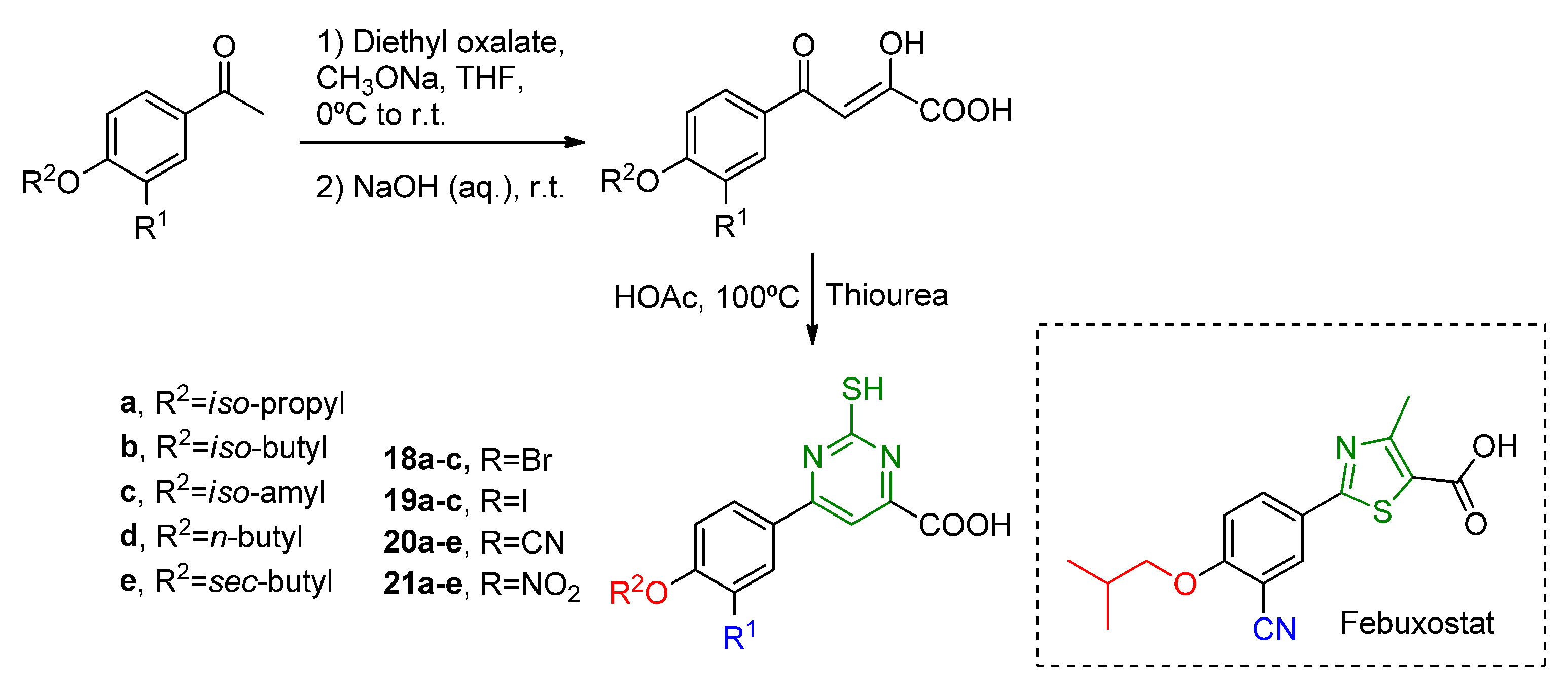
Febuxostat analogs have also been explored with a six-member heterocycle instead of the five-member. This is the case of 2-mercapto-6-phenylpyrimidine-4-carboxylic acid derivatives
18a–
c,
19a–
c,
20a–
e,
21a–
e synthesized by Shi and coworkers (
Scheme 10) [
26]. The thiazole ring was replaced by a mercapto-pyrimidine moiety, and the inhibition activity was compared to febuxostat (
Table 7). It was observed that the most promising compound (
20b) had a cyano group at R
1 and an
iso-butyl substituent at R
2, presenting an in vitro IC
50 of 132 nM. However, the inhibition potency was approximately ten times lower than febuxostat (in vitro IC
50 = 13 nM). The other derivatives retained the nanomolar potency, but presented even higher in vitro IC
50 values. SAR showed that the cyano group is more promising than other electron-withdrawing substituents such as nitro, iodine, and bromide. However, as foreseen by molecular docking, changes in the structure show that the cyano group from phenyl moiety does not properly interact with the XO active site, whereas the mercapto group interacts via a hydrogen bond with Mos4004 and protonated Glu802 residue. Kinetic assays revelated the mixed type inhibition mechanism of
20b, similar to febuxostat and Y-700.
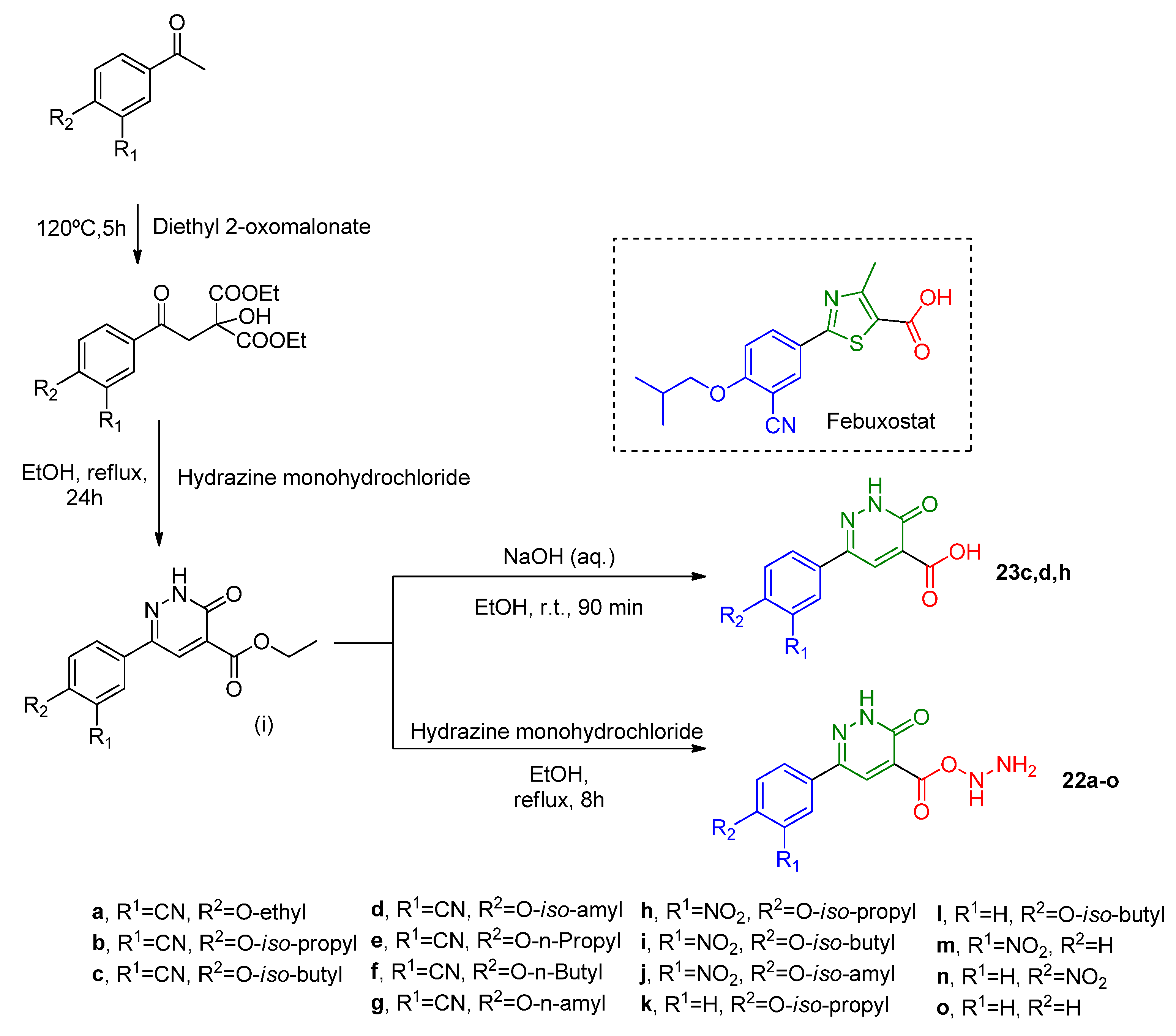
Using a similar strategy, Zhang and coworkers [
46] synthesized two classes of pyridazine derivatives by replacement of the thiazole group of febuxostat with a six-member heterocycle moiety. The first two steps of the strategic synthesis were similar for both classes of compounds. The procedure followed the hydrazinolysis pathway to obtain the carbohydrazide derivatives
22a–
o or the hydrolysis pathway to obtain the carboxylic acid derivatives
23c,
d,
h (
Scheme 11). Surprisingly, the carboxylic acid derivatives
23c,
d,
h were not active, while the carbohydrazide derivatives
22a–
o presented lower in vitro IC
50 than allopurinol (in vitro IC
50 = 6.43 μM), but much higher than febuxostat (in vitro IC
50 = 0.018 μM) (
Table 8). The difference in the two classes of compounds activity was explained through molecular docking analysis of analog structures,
6-(3-Cyano-4-isobutoxyphenyl)-3-oxo-2,3-dihydropyridazine-4- carbohydrazide (
22c) and
6-(3-Cyano-4-isobutoxyphenyl)-3-oxo-2,3-dihydropyridazine-4-carboxylic acid (
23c). According to this analysis, more prominent interactions were found between the carbohydrazide group with the XO active site than a carboxylic acid. The carbohydrazide group of
23c forms hydrogens bounds with Ag880, Glu802, and Mos3004 residues, while the carboxylic acid of
22c only interacts with Glu802. SAR analysis related to the R
1 and R
2 phenyl substituents showed that both cyano and nitro groups at R
1 with O-iso-propyl at R
2 are beneficial for inhibition activity (IC
50 of 1.03 μM and 1.92 μM, for
22b and
22h, respectively). Kinetic studies comprising the most potent compound (
22b) pointed out a mixed-type inhibition mechanism.
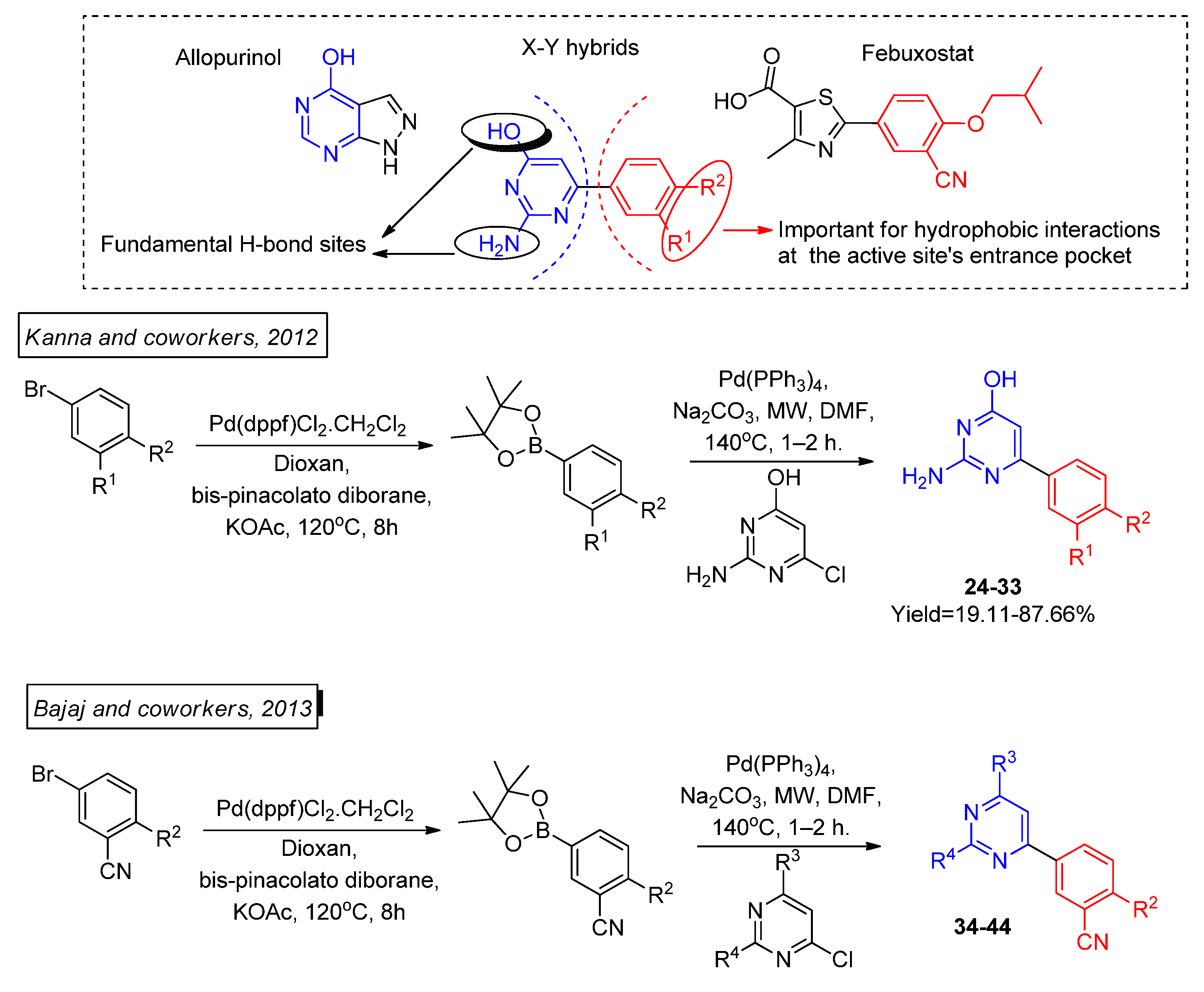
Guided by SAR studies, Khanna and coworkers managed to successfully plan diverse structural modification in the isocytosine scaffold [
47], which was further explored by the same research group by synthesizing and testing novel molecular deviations [
48]. Isocytosine was earlier recognized as a promising lead compound to be explored as a XO inhibitor by the group’s virtual screening collection. More than thirty novel allopurinol and febuxostat hybrid compounds, of which we highlight the most promising ones,
24–
32 and
33–
43 (
Scheme 12), were organized by structural similarity to detach the SAR influence in modulating the biological activity (
Table 9), with extremely low values of in vitro IC
50 compared to the control drugs allopurinol and febuxostat. Moreover,
29,
30,
32, and
43 presented good in vivo efficacy in a hyperuricemic rat model compared to allopurinol. The conjugation of allopurinol and febuxostat molecular fractions might explain the promising results consisting of lead compounds where the -NH
2 and -OH groups of isocytosine formed fundamental hydrogen bonds with the target’s active site in limited SAR studies, while the R
1 and R
2 substituents played a key role by managing hydrophobic interactions at the active site pocket’s entrance by both electrostatic interactions and steric effects.
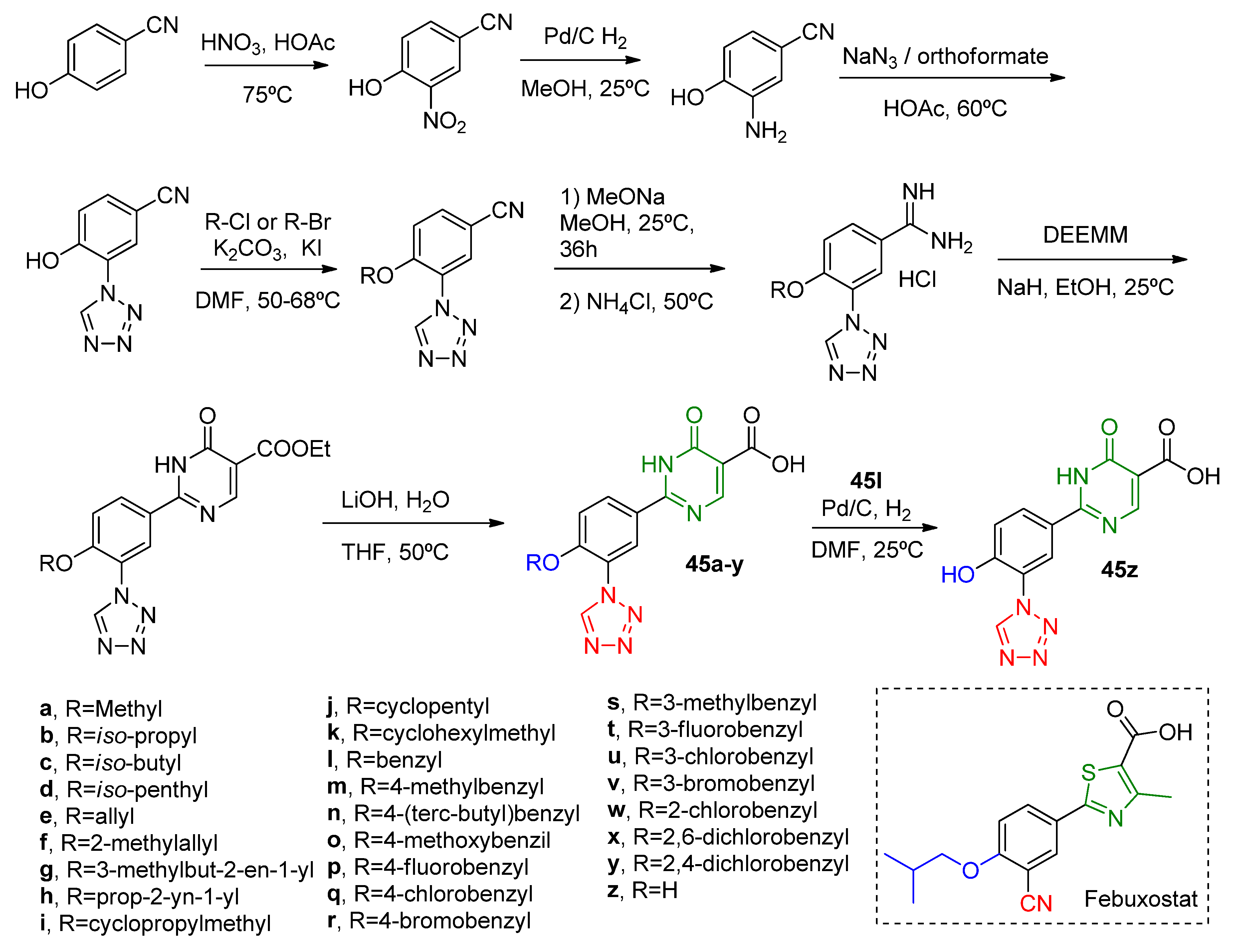
A multi-step synthesis of febuxostat–allopurinol hybrids
45a–
z (
Scheme 13) was made by Zhang and coworkers [
49] to explore the isosteric replacement of the cyano group of the above-discussed works molecules [
47,
48] and execute a SAR analysis. XO inhibition was evaluated through IC
50 analysis that, in general, had values comparable to febuxostat (
Table 10). The most active compounds had
meta-chlorobenzyl or
meta-bromobenzyl anchored at R with in vitro IC
50 of 28.8 nM (
45u) and 45.0 nM (
45v). A SAR analysis showed that hydrophobic compounds at R are essential for a good inhibition potential. Docking studies of the most potent inhibitor (
45u) showed that the tetrazole ring can be accommodated at the XO active site and form hydrogen bonds with Asn768 and Lys771; additionally, the carboxyl group of pyrimidine also interacts with Ag880 and Mo-OH residues. The six-membered heterocycle group of
45u shows the same aromatic interactions as the thiazole group in febuxostat, while chlorobenzyl displays a π–π interaction with Phe649. Lastly, in vivo analysis demonstrated that oral administration of a single dose (5 mg/kg) of
45u in hyperuricemic rats, by administration of potassium oxonate, can lower the serum levels of UA.
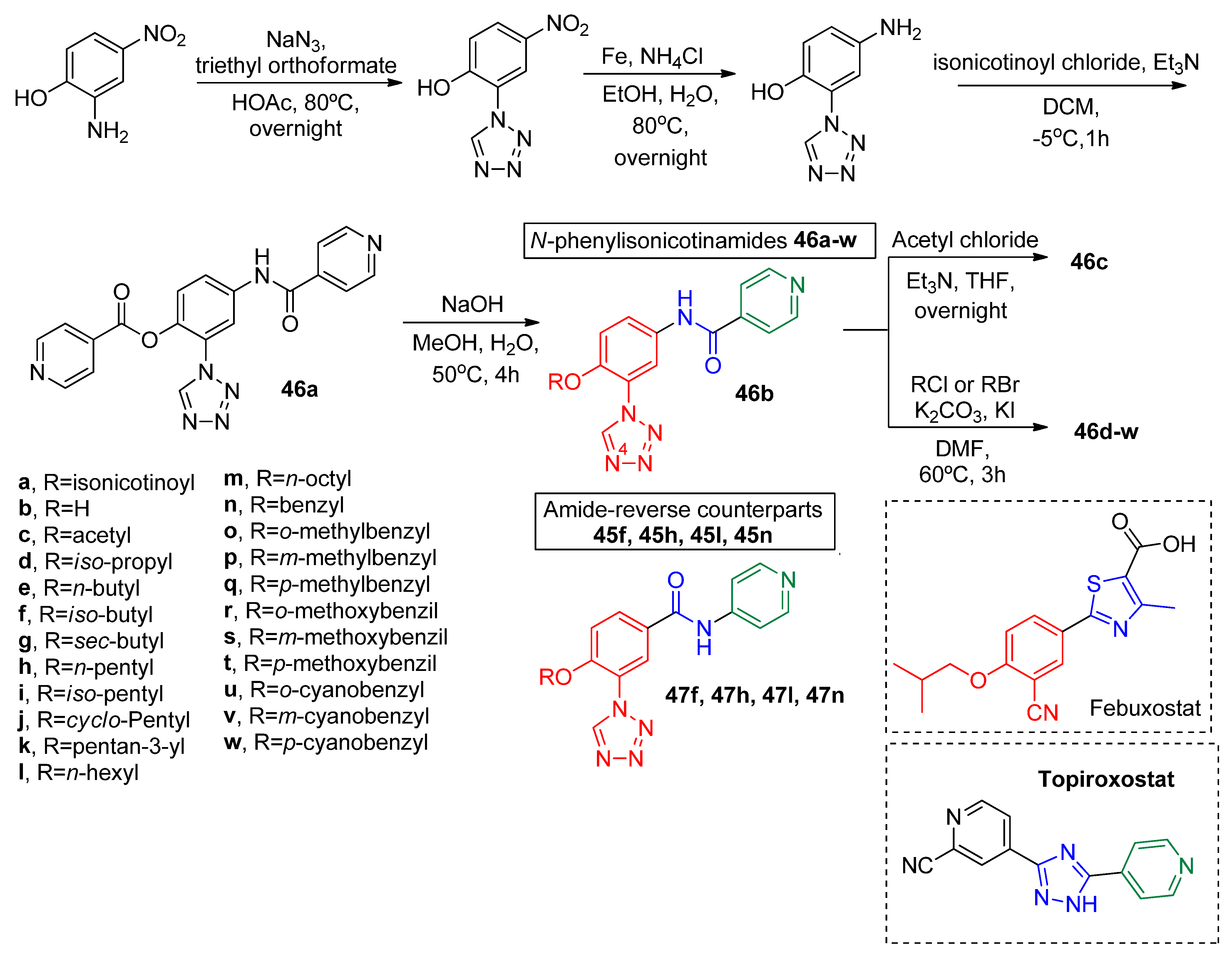
A complementary work by Zhang and coworkers further explored the bioisosteric replacement of the febuxostat cyano group by the 1,2,3,4-tetrazole [
50]. The synthesized derivative
46a–
w (
Scheme 14) hybrids based in topiroxostat and febuxostat molecular scaffold presented potency in the nanomolar range (IC
50 between 31 nM and 603 nM), except for
46a–
b, which were inactive (
Table 11). The general R group influence trend over activity modulation was that increasing hydrophobicity and volume led to better XO inhibition. Additionally, aromatic groups containing electron-withdrawing components at a meta position (
46s and
46v) seemed to be the most beneficial pattern to obtain high potency derivatives. Moreover, they further synthesized the analogs
47f,
h,
l,
n. Differently from
46a–
w, where the cyanophenyl portion was bonded to the nitrogen atom of the amide linker, the N-phenylisonicotinamide counterparts
47f,
h,
l,
n ended up being over twenty times less active (
Table 11). A molecular docking study and a molecular dynamics simulation for the most active hybrid
46v revealed a strong interaction with the XO’s active site as tetrazole N
4 nitrogen and Asn768; pyridine
para-N and Glu1261; cyano group and Ser876 (two H-bonds at the same time, one by the amino acid OH group and the other by NH group); and carbonyl group with Arg880 and Thr1010 via a water bridge. Lastly, kinetic studies involving the selected hybrid
46v revealed a mixed-type of inhibition mechanism.
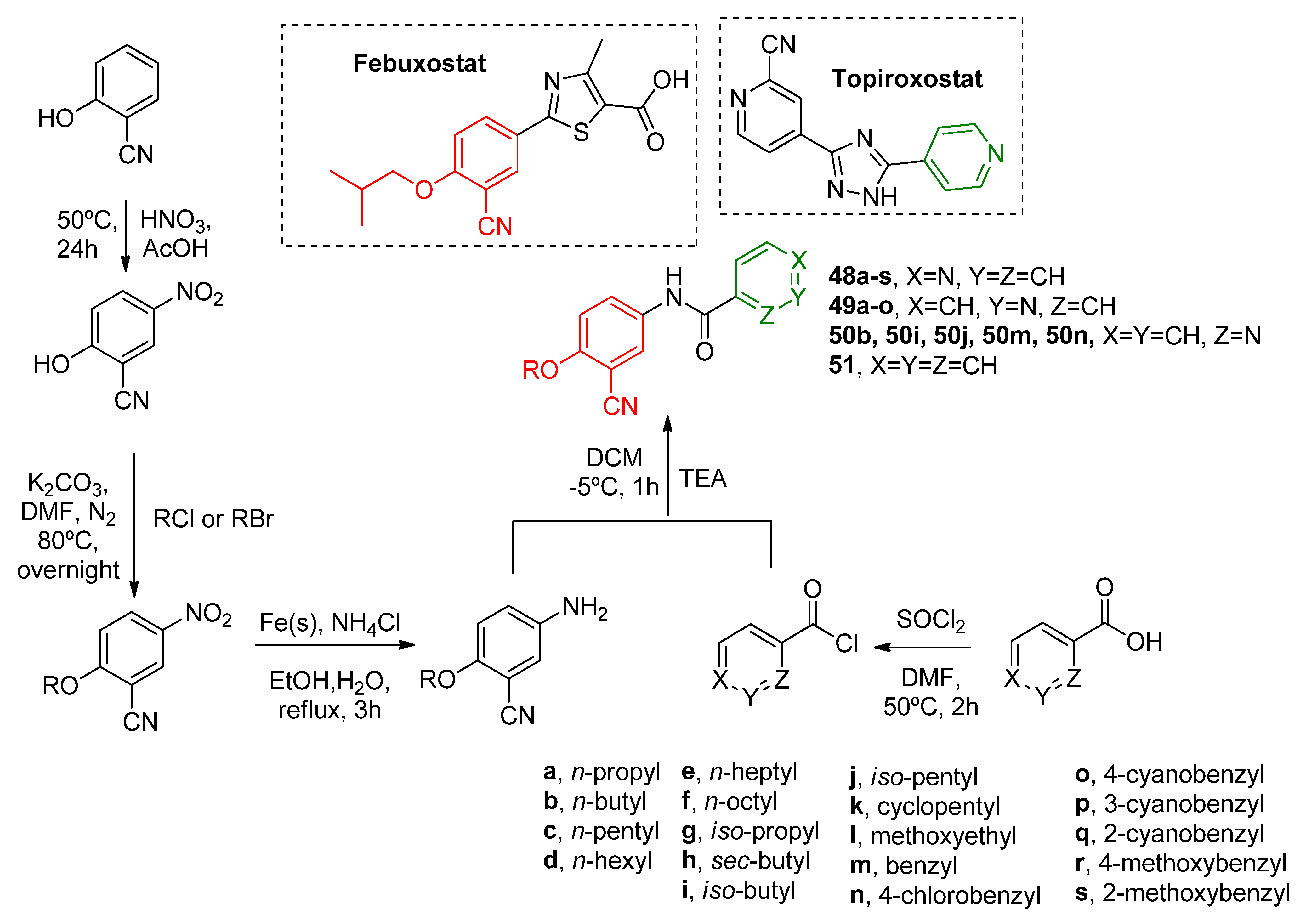
Motivated to obtain different hybrid derivatives based in topiroxostat and febuxostat molecular structures, Zhang and coworkers [
51] explored hybrid structures of isonicotinamide (
48a–
s), nicotinamide (
49a–
o), picolinamide (
50b,
i,
j,
m,
n), and benzamide (
51) derivatives where the pyridine moiety nitrogen position also varied (
Scheme 15). This time using an amide linker as an isosteric replacement for the previously discussed imidazole linker. SAR showed that large linear carbon chains lead to reduced potency derivatives (
Table 12), as observed by the increase in IC
50 values from
48b (in vitro IC
50 = 2.3 μM) to
48f (in vitro IC
50 = 19.2 μM). Additionally,
orto- (
49a–
o),
meta-N (
50b,
i,
j,
m,
n) and N absence (
51) lead mostly to unactive derivatives. On the other hand, aromatic R groups in
para-N derivatives furnished some of the most potent inhibitors, such as
48q with in vitro IC
50 of 0.3 μM and
48r with in vitro IC
50 of 0.6 μM. Overall, sixteen out of the nineteen isonicotinamide derivatives were more potent than allopurinol (in vitro IC
50 = 8.5 μM), but less active than febuxostat (in vitro IC
50 = 0.015 μM). As observed through molecular docking studies, isonicotinamide
para-N formed a fundamental H-bond with Glu1261 residue. Moreover, regarding the amide group, pronounced interactions with Thr1010, Arg880, and Glu802 were observed, distancing the cyano group from Asn768, which probably lead to the decreased potency of isonicotinamide derivatives compared to febuxostat and its analogs. Lastly, kinetic studies revealed
48q as a mixed-type inhibitor of XO. Which might be explained by potent inhibition of oxidized and reduced forms of XO.
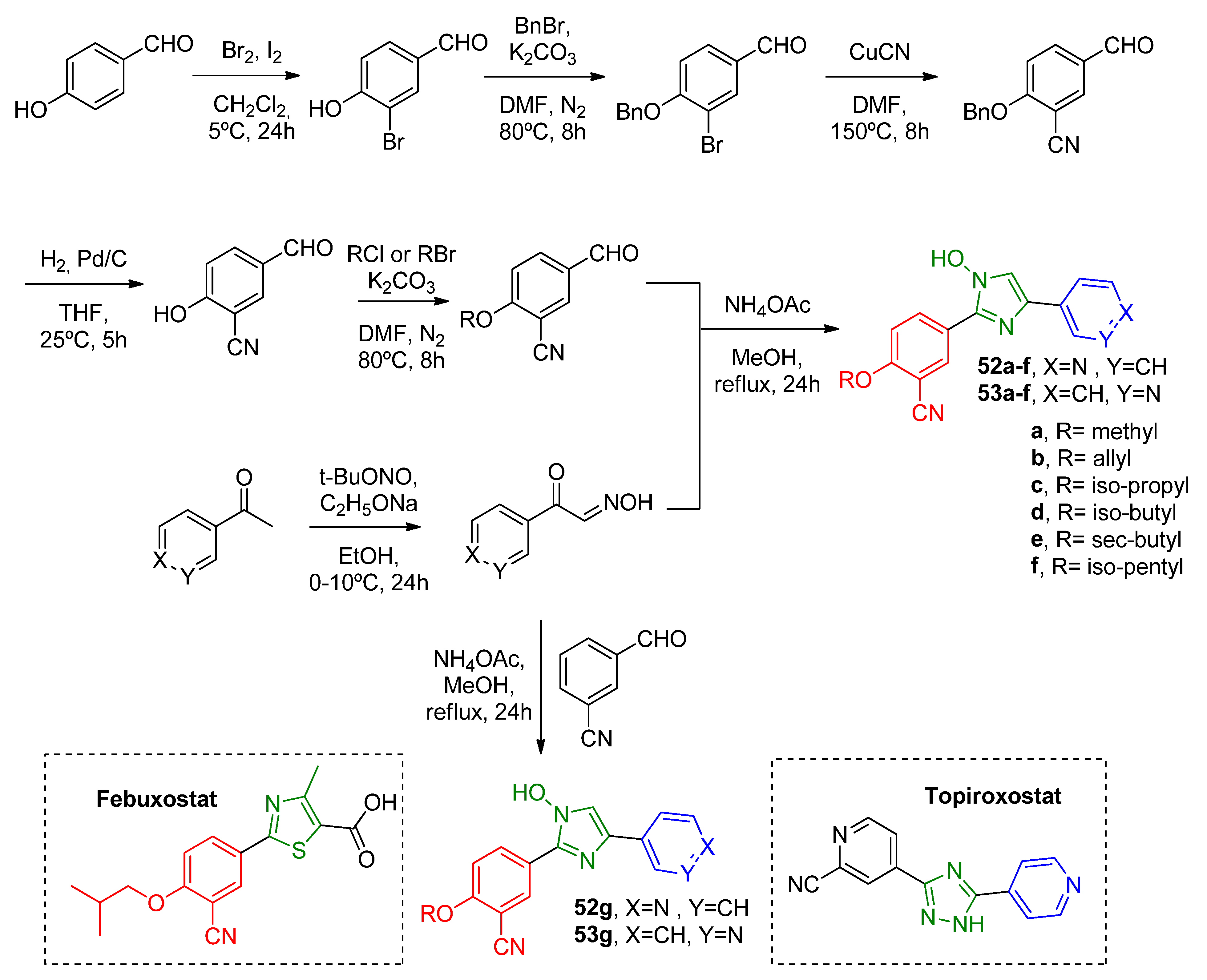
Under a similar approach, Zhang and coworkers [
52] developed two novel families of isosteric compounds comprising a cyanophenyl-imidazole scaffold differently connected to a pyridine ring (
Scheme 16). The novel synthesized topiroxostat-based
para-substituted pyridine (
52a–
g) and
meta-substituted pyridine (
53a–
g) also had an R sidechain variated with different hydrocarbon substituents for SAR investigation (
Table 13). A SAR analysis pointed out that the steric effect of bulkier substituents contributed to better activity. Further, a 3D-QSAR model with the Topomer comparative molecular field analysis method was used to understand the interactions between
52f/53f and the XO active site. It was observed that pyridine with
para-N forms a hydrogen bond with Glu1261 residue, while
meta-N does not show any interaction, which could explain the great activity modulation between the isosteric compound families. Considering the aforementioned, the more pronounced activity of compound
52f (in vitro IC
50 = 0.64 μM) could be understood. However, it was still not as active as topiroxostat (in vitro IC
50 = 0.0048 μM). Lastly, kinetic studies involving
52f showed its mixed-type inhibition mechanism on XO.
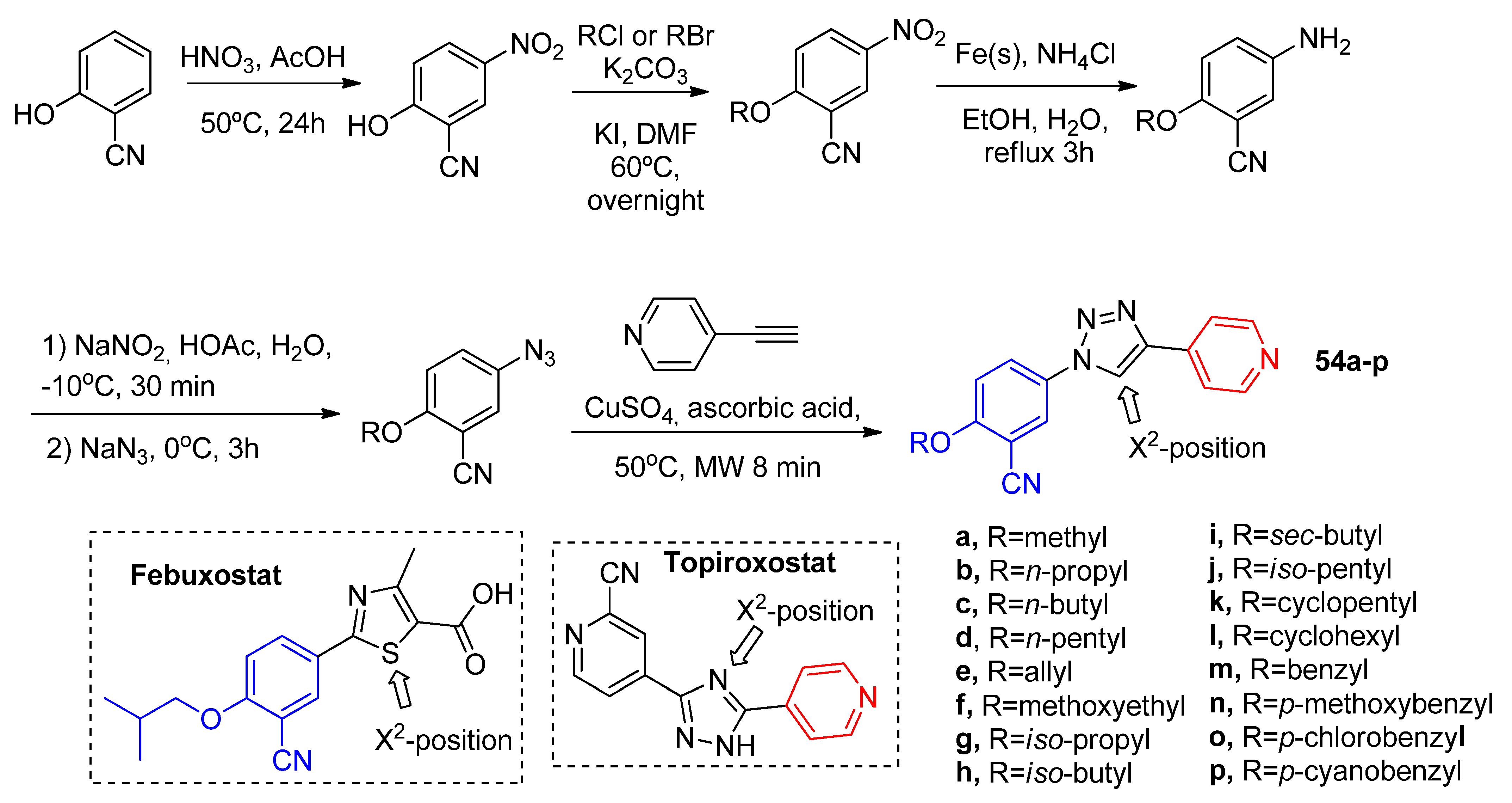
Aiming an isosteric replacement of the amide linker by an 1,2,3-triazole nucleus, Zhang and coworkers [
53] synthesized the novel febuxostat–topiroxostat hybrid derivatives
54a–
p (
Scheme 17) correlated with Zhang and coworkers’ previous work [
50]. These compounds presented a similar inhibition of XO to allopurinol, but lower than febuxostat (
Table 14). The most potent compounds were those with
iso-pentyl (
54j) and cyclopentyl (
54k) tails as substituents with IC
50 of 8.1 μM and 6.7 μM, respectively. Docking analysis of
54k reveals similar interactions of topiroxostat at the XO active site, including a lipophilic interaction of the cyclopentyloxy ether tail with Leu648, Phen649, and Phen1013 residues. Was also observed that substitution of the N atom at X
2-position for a -CH was not favorable due to the distance increase of the triazole ring from the Glu802 residue.


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















