A Review of Enzyme Induced Carbonate Precipitation (EICP): The Role of Enzyme Kinetics





Abstract
:1. Introduction
2. Biogeochemical Reactions in EICP
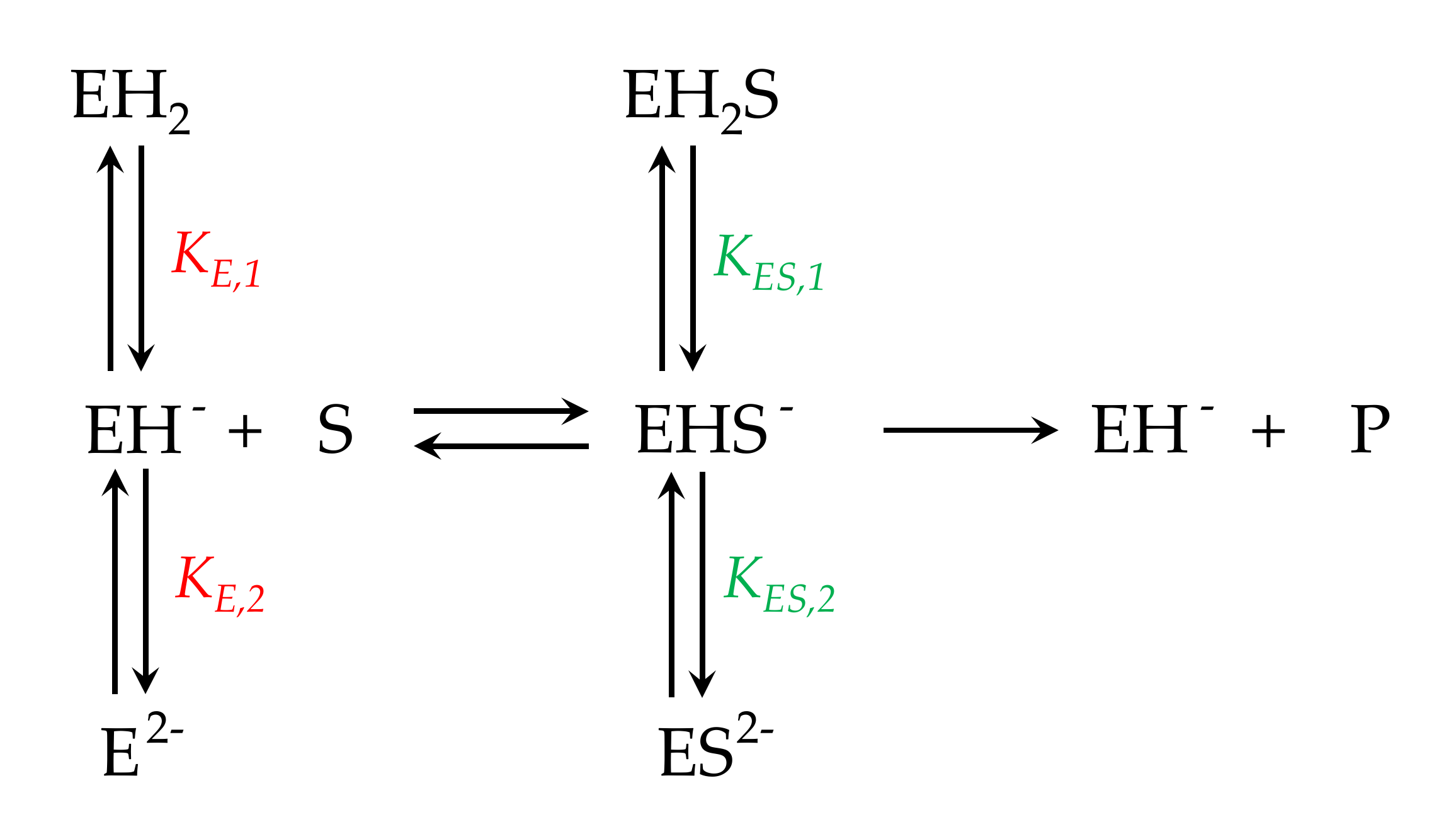
2.1. Molecular Structure of Urease Enzyme
2.2. Urease Catalysed Chemical Reactions
3. Classical Enzyme Kinetics and Ureolytic Catalysis
3.1. Historical Development of Enzyme Kinetics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Historical Achievement | Reference |
|---|---|---|
| 1913 | Proposed the basic description of enzymatic reactions. | Michaelis and Menten [10] |
| 1955 | Stated that for QSSA, there should be an excess in substrate concentration. | Laidler [42] |
| 1962 | Observed that QSSA may not hold for a reaction with large reverse bimolecular velocity. | Hommes [43] |
| 1965 | Stated that a brief transient state is required for QSSA. | Wong [44] |
| 1979 | Found that for QSSA to hold, the substrate/enzyme ratio should be >100. | Stayton and Fromm [45] |
| 1997 | Developed a closed-form solution for the basic enzyme-substrate reaction. | Schnell and Mendoza [47] |
| 2003 | Redeveloped a closed-form solution at high enzyme concentrations. | Tzafriri [50] |
| Since 2009 | Development of various computer programs to solve the integrated Michaelis-Menten equation. | Johnson [12], Kuzmič [51], Zavrel, Kochanowski [53] |
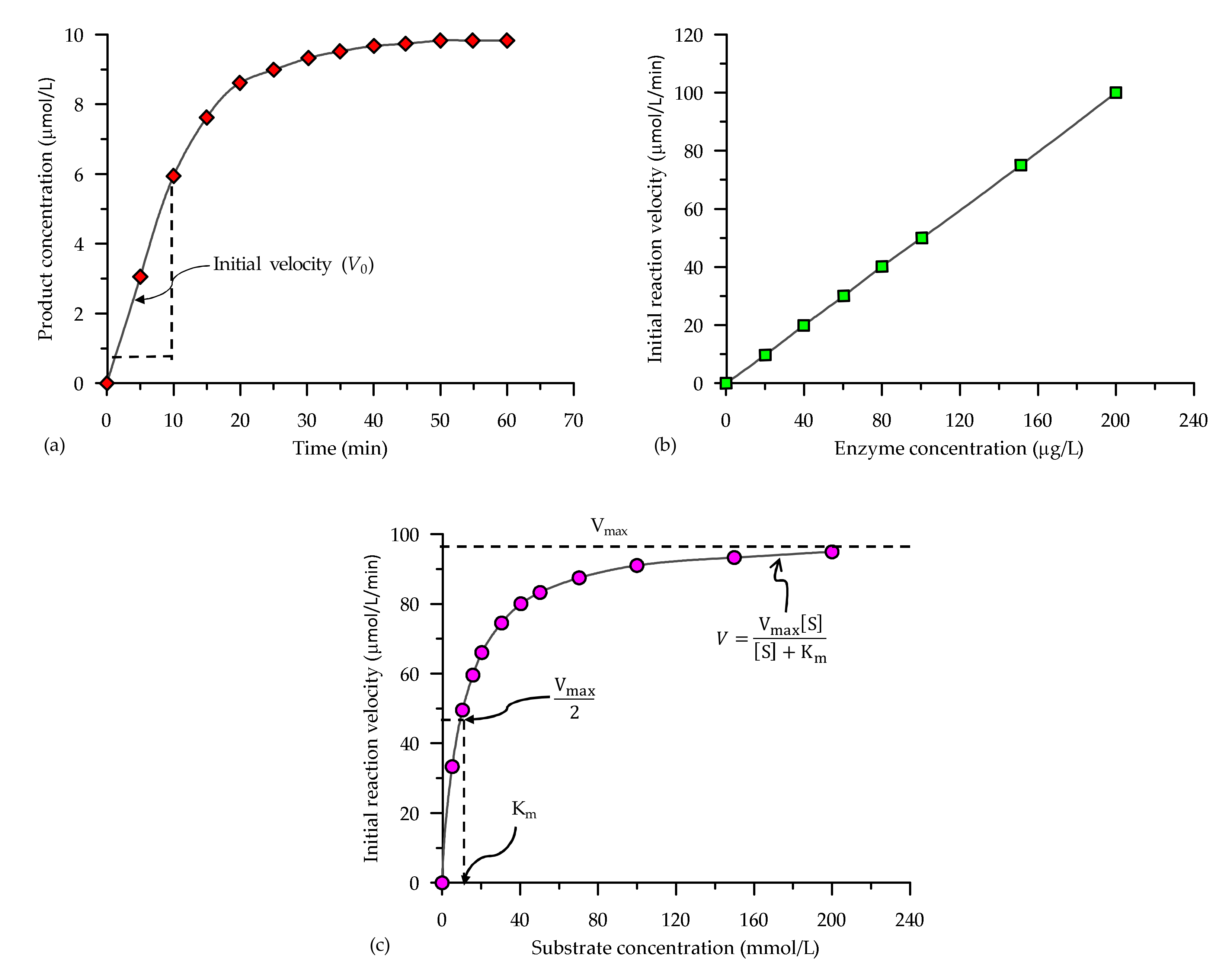
3.2. Enzyme-Catalysed Reaction Methods
3.3. The Michaelis-Menten Equation
4. Estimation of Kinetic Parameters
4.1. Integrated Michaelis–Menten Rate Equations
4.2. Closed-Form Solution of the Rate Equation
5. Applications of Enzyme Kinetic Models in EICP
5.1. Factors Affecting the Kinetic Parameters
5.1.1. pH
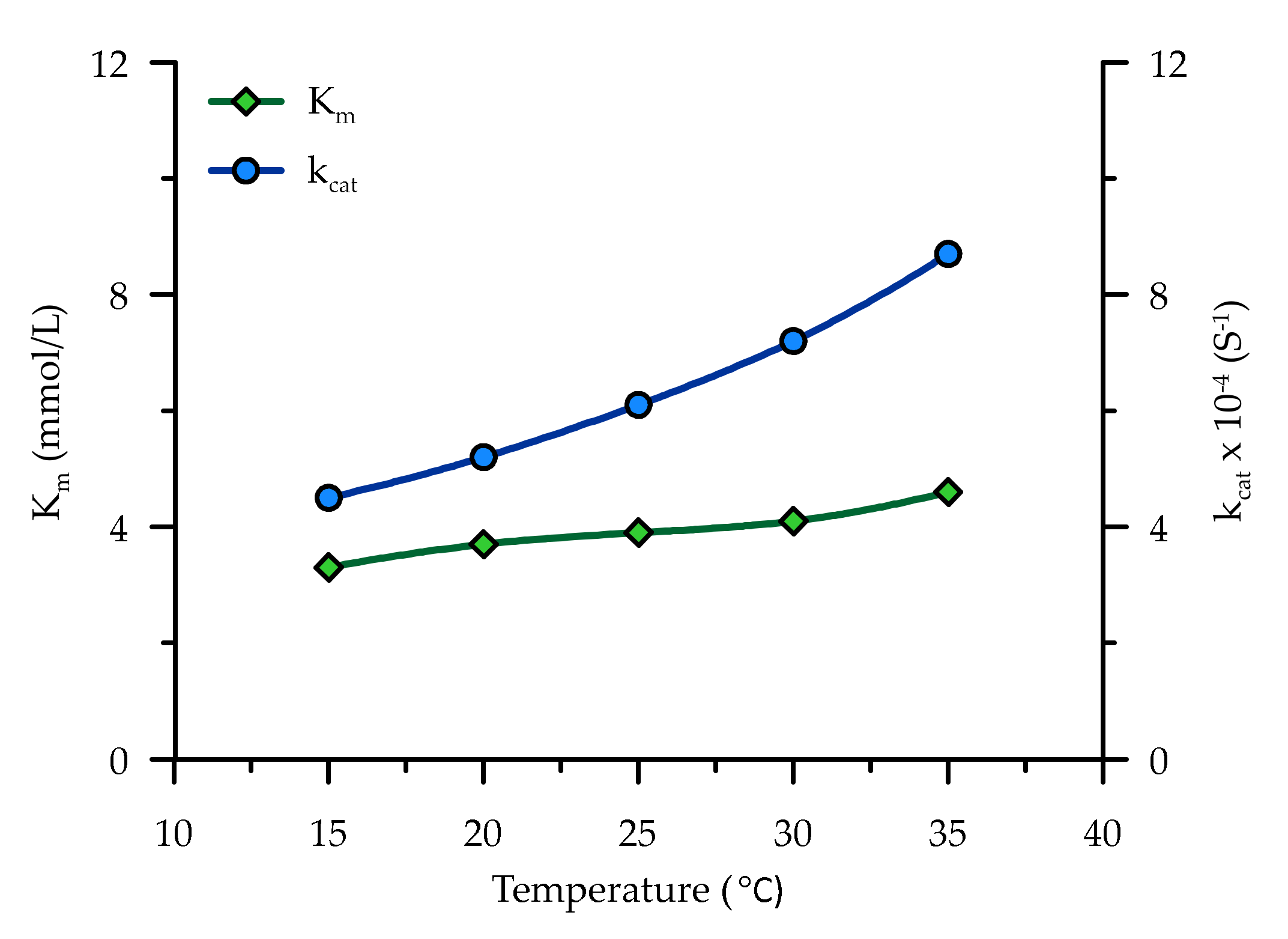
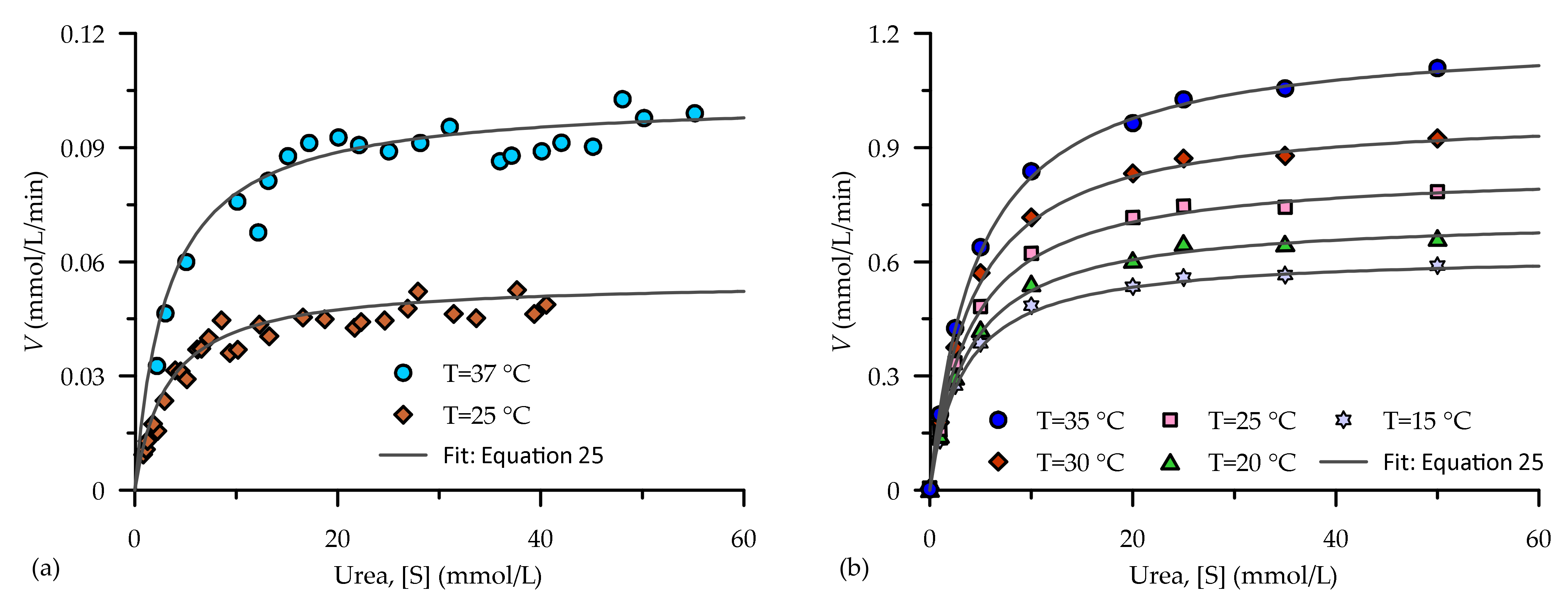
5.1.2. Temperature
5.1.3. Product Inhibition
5.2. Proposed Kinetic Model for EICP
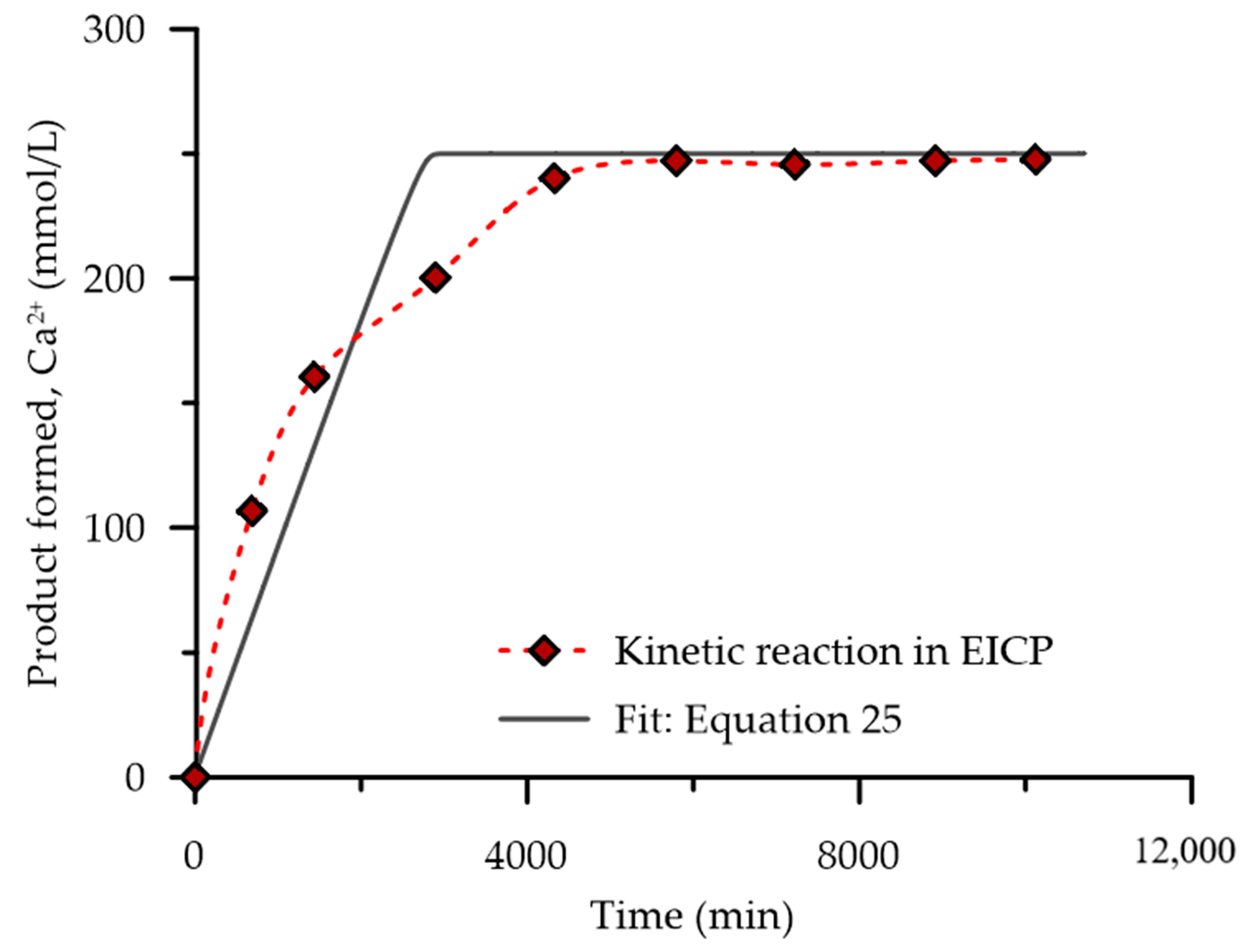
5.3. Evaluation of the Proposed Kinetic Equation
6. Urease Inhibition
7. Engineering Applications of Urease Aided- Precipitation
7.1. Improvement of the Strength and Stiffness of Soils
7.2. Erosion and Dust Control
7.3. Removal of Heavy Metals
8. Conclusions and Future Perspectives
- The activity of the urease enzyme is largely controlled by the presence of a binuclear Ni complex active site in the β-sheet structure and the dynamic opening and closing of the mobile flap located adjacent to the active site.
- Studies on optimisation of the EICP process have often been conducted by using the discontinuous approach, which involves mixing the substrate and enzyme and measuring the product formed after a set period. However, this approach cannot easily capture the catalytic properties, such as the influence of urease activity and product inhibition on the enzyme-catalysed reaction. Therefore, the continuous enzyme kinetic assay, which involves mixing the enzyme with the substrate and continuously measuring the product formed or the dissociation of the substrate over time, should be considered in future studies.
- It is understood from this study that the reaction velocity of an enzyme catalysed reaction is mainly influenced by pH, temperature and inhibitors (ammonium ion). A meta-analysis of data from a previous study indicate that pH and ammonium ions greatly affect compared to , whereas was greatly influenced by temperature. A modified form of the Michaelis–Menten equation was proposed in this study, which can be used to capture the kinetic reaction in EICP under various conditions.
- The findings from this study indicate that ignoring the influence of product inhibition in an enzyme-catalysed reaction may result in a poor prediction of the kinetic parameters. Hence, various sources of urease inhibitors including amides and esters of phosphoric acid, thiols, hydroxamic acids, phosphinic and thiophosphinic acids, boric acid, phosphate, heavy metal ions, bismuth compounds, quinones and fluoride have been studied.
- Although the kinetic equations analysed and proposed in this study are useful for the EICP process, future studies on the influence of enzyme kinetic reactions in different soil environments are highly recommended. The development of kinetic models that capture the effects of using an enzyme from different plant sources should also be considered for future studies.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Notations
| maximal reaction velocity | |
| Michaelis constant | |
| V | reaction velocity |
| V0 | initial reaction velocity |
| S | substrate |
| E | enzyme |
| P | product |
| ES | enzyme-substrate complex |
| EP | enzyme-product complex |
| rate constant for the formation of the ES complex | |
| rate constant for the dissociation of the ES complex | |
| catalytic rate constant or a turnover number | |
| original concentration of urease enzyme | |
| original concentration of substrate | |
| [S] | initial concentration of substrate |
| initial concentration of product | |
| t | reaction time |
| apparent product inhibition constant | |
| specificity constant | |
| Lambert function | |
| molecular dissociation constants for the free enzyme | |
| molecular dissociation constants for the enzyme-substrate complex | |
| initial | |
| activation energy | |
| R | gas constant |
| temperature at which = 1 min−1 | |
| apparent product inhibition constant | |
| ammonium ion concentration |
References
- Krajewska, B. Urease-aided calcium carbonate mineralization for engineering applications: A review. J. Adv. Res. 2017, 13, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Putra, H.; Yasuhara, H.; Kinoshita, N. Applicability of natural zeolite for nh-forms removal in enzyme-mediated calcite precipitation technique. Geosciences 2017, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Ahenkorah, I.; Rahman, M.M.; Karim, M.R.; Teasdale, P.R. A comparison of mechanical responses for microbial and enzyme-induced cemented sand. Géotechnique Lett. 2020, 10, 1–26. [Google Scholar] [CrossRef]
- Neupane, D.; Yasuhara, H.; Kinoshita, N.; Ando, Y. Distribution of mineralized carbonate and its quantification method in enzyme mediated calcite precipitation technique. Soils Found. 2015, 55, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, N.M. Applications of Enzyme Induced Carbonate Precipitation (Eicp) for Soil Improvement. Ph.D. Thesis, Arizona State University, Tempe, AZ, USA, 2015. [Google Scholar]
- Dilrukshi, R.; Kawasaki, S. Effective use of plant-derived urease in the field of geoenvironmental. Geotech. Eng. J. Civ. Environ. Eng. 2016, 6, 2. [Google Scholar]
- Suárez, D.; Díaz, N.; Merz, K.M. Ureases: Quantum chemical calculations on cluster models. J. Am. Chem. Soc. 2003, 125, 15324–15337. [Google Scholar] [CrossRef]
- Zimmer, M. Are classical molecular mechanics calculations still useful in bioinorganic simulations? Coord. Chem. Rev. 2009, 253, 817–826. [Google Scholar] [CrossRef]
- Smyj, R.P. A conformational analysis study of a nickel (ii) enzyme: Urease. J. Mol. Struct. THEOCHEM 1997, 391, 207–223. [Google Scholar] [CrossRef]
- Michaelis, L.; Menten, M.L. Die kinetik der invertinwirkung. Biochem. Z 1913, 49, 333–369. [Google Scholar]
- Krajewska, B. Ureases i. Functional, catalytic and kinetic properties: A review. J. Mol. Catal. B Enzym. 2009, 59, 9–21. [Google Scholar] [CrossRef]
- Johnson, K.A. Fitting enzyme kinetic data with kintek global kinetic explorer. Methods Enzymol. 2009, 467, 601–626. [Google Scholar]
- Krajewska, B. A combined temperature-ph study of urease kinetics. Assigning pka values to ionizable groups of the active site involved in the catalytic reaction. J. Mol. Catal. B Enzym. 2016, 124, 70–76. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Ciurli, S.; Mangani, S. A new proposal for urease mechanism based on the crystal structures of the native and inhibited enzyme from bacillus pasteurii: Why urea hydrolysis costs two nickels. Structure 1999, 7, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Krajewska, B. Ureases. Ii. Properties and their customizing by enzyme immobilizations: A review. J. Mol. Catal. B Enzym. 2009, 59, 22–40. [Google Scholar] [CrossRef]
- Ahenkorah, I.; Rahman, M.M.; Karim, M.R.; Teasdale, P.R. Optimization of Enzyme Induced Carbonate Precipitation (Eicp) as a Ground Improvement Technique; Geo-Congress 2020: Foundations, Soil Improvement, and Erosion; American Society of Civil Engineers: Reston, VA, USA, 2020; pp. 552–561. [Google Scholar]
- Neupane, D.; Yasuhara, H.; Kinoshita, N.; Unno, T. Applicability of enzymatic calcium carbonate precipitation as a soil-strengthening technique. J. Geotech. Geoenviron. Eng. 2013, 139, 2201–2211. [Google Scholar] [CrossRef]
- Almajed, A.; Khodadadi Tirkolaei, H.; Kavazanjian, E., Jr. Baseline investigation on enzyme-induced calcium carbonate precipitation. J. Geotech. Geoenviron. Eng. 2018, 144, 04018081. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N.; Neupane, D. Optimization of Calcite Precipitation as a Soil Improvement Technique. In Proceedings of the 2nd Makassar International Conference on Civil Engineering, Makassar, Indonesia, 11–12 August 2015; pp. 11–12. [Google Scholar]
- Carmona, J.P.; Oliveira, P.J.V.; Lemos, L.J. Biostabilization of a sandy soil using enzymatic calcium carbonate precipitation. Procedia Eng. 2016, 143, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Sumner, J.B. The isolation and crystallization of the enzyme urease preliminary paper. J. Biol. Chem. 1926, 69, 435–441. [Google Scholar] [CrossRef]
- Mazzei, L.; Musiani, F.; Ciurli, S. The structure-based reaction mechanism of urease, a nickel dependent enzyme: Tale of a long debate. J. Biol. Inorg. Chem. 2020, 25, 829–845. [Google Scholar] [CrossRef]
- Kafarski, P.; Talma, M. Recent advances in design of new urease inhibitors: A review. J. Adv. Res. 2018, 13, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Follmer, C. Insights into the role and structure of plant ureases. Phytochemistry 2008, 69, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.; Island, M.D.; Hausinger, R.P. Molecular biology of microbial ureases. Microbiol. Rev. 1995, 59, 451–480. [Google Scholar] [CrossRef]
- Sirko, A.; Brodzik, R. Plant ureases: Roles and regulation. Acta Biochim. Pol. 2000, 47, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, N.; Kayastha, A.M.; Srivastava, P.K. Purification and characterization of urease from dehusked pigeonpea (cajanus cajan l.) seeds. Phytochemistry 2002, 61, 513–521. [Google Scholar] [CrossRef]
- Nam, I.-H.; Chon, C.-M.; Jung, K.-Y.; Choi, S.-G.; Choi, H.; Park, S.-S. Calcite precipitation by ureolytic plant (canavalia ensiformis) extracts as effective biomaterials. KSCE J. Civ. Eng. 2015, 19, 1620–1625. [Google Scholar] [CrossRef]
- Takatsu, M.; Nadeeka, R.; Kawasaki, S. Development of biogrouting using plant-derived urease and calcium phosphate compound. In Proceedings of the 50th US Rock Mechanics/Geomechanics Symposium, Huston, TX, USA, 26–29 June 2016. [Google Scholar]
- Balasubramanian, A.; Ponnuraj, K. Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J. Mol. Biol. 2010, 400, 274–283. [Google Scholar] [CrossRef]
- Kunduru, K.R.; Kutcherlapati, S.R.; Arunbabu, D.; Jana, T. Armored urease: Enzyme-bioconjugated poly (acrylamide) hydrogel as a storage and sensing platform. Methods Enzymol. 2017, 590, 143–167. [Google Scholar]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, B.; Zaborska, W. Jack bean urease: The effect of active-site binding inhibitors on the reactivity of enzyme thiol groups. Bioorg. Chem. 2007, 35, 355–365. [Google Scholar] [CrossRef]
- Roberts, B.P.; Miller, B.R., III; Roitberg, A.E.; Merz, K.M., Jr. Wide-open flaps are key to urease activity. J. Am. Chem. Soc. 2012, 134, 9934–9937. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Mandani, S.; Sarma, T.K. Biogenic growth of alloys and core-shell nanostructures using urease as a nanoreactor at ambient conditions. Sci. Rep. 2013, 3, 2601. [Google Scholar] [CrossRef] [Green Version]
- Kucharski, E.S.; Cord-Ruwisch, R.; Whiffin, V.; Al-thawadi, S.M. Microbial Biocementation; Google Patents: San Francisco, CA, USA, 2012. [Google Scholar]
- Kappaun, K.; Piovesan, A.R.; Carlini, C.R.; Ligabue-Braun, R. Ureases: Historical aspects, catalytic, and non-catalytic properties–A review. J. Adv. Res. 2018, 13, 3–17. [Google Scholar] [CrossRef]
- Dixon, N.E.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack bean urease (ec 3.5. 1.5). Metalloenzyme. Simple biological role for nickel. J. Am. Chem. Soc. 1975, 97, 4131–4133. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, M. Molecular mechanics evaluation of the proposed mechanisms for the degradation of urea by urease. J. Biomol. Struct. Dyn. 2000, 17, 787–797. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular biology of the cell 4th edn (new york: Garland science). Ann. Bot. 2002, 91, 401. [Google Scholar]
- Laidler, K.J. Theory of the transient phase in kinetics, with special reference to enzyme systems. Can. J. Chem. 1955, 33, 1614–1624. [Google Scholar] [CrossRef]
- Hommes, F. The integrated michaelis-menten equation. Arch. Biochem. Biophys. 1962, 96, 28–31. [Google Scholar] [CrossRef]
- Wong, J.T.-F. On the steady-state method of enzyme kinetics. J. Am. Chem. Soc. 1965, 87, 1788–1793. [Google Scholar] [CrossRef]
- Stayton, M.M.; Fromm, H.J. A computer analysis of the validity of the integrated michaelis-menten equation. J. Theor. Biol. 1979, 78, 309–323. [Google Scholar] [CrossRef]
- Beal, S.L. On the solution to the michaelis-menten equation. J. Pharmacokinet. Biopharm. 1982, 10, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Schnell, S.; Mendoza, C. Closed form solution for time-dependent enzyme kinetics. J. Theor. Biol. 1997, 187, 207–212. [Google Scholar] [CrossRef]
- Corless, R.M.; Gonnet, G.H.; Hare, D.E.; Jeffrey, D.J.; Knuth, D. Lambert’s w function in maple. Maple Tech. Newsl. 1993, 9, 12–22. [Google Scholar]
- Fritsch, F.N.; Shafer, R.; Crowley, W. Solution of the transcendental equation wew= x. Commun. ACM 1973, 16, 123–124. [Google Scholar] [CrossRef]
- Tzafriri, A.R. Michaelis-menten kinetics at high enzyme concentrations. Bull. Math. Biol. 2003, 65, 1111–1129. [Google Scholar] [CrossRef]
- Kuzmič, P. Dynafit—A software package for enzymology. Methods Enzymol. 2009, 467, 247–280. [Google Scholar]
- Bevc, S.; Konc, J.; Stojan, J.; Hodošček, M.; Penca, M.; Praprotnik, M.; Janežič, D. Enzo: A web tool for derivation and evaluation of kinetic models of enzyme catalyzed reactions. PLoS ONE 2011, 6, e22265. [Google Scholar] [CrossRef]
- Zavrel, M.; Kochanowski, K.; Spiess, A.C. Comparison of different approaches and computer programs for progress curve analysis of enzyme kinetics. Eng. Life Sci. 2010, 10, 191–200. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N.; Hirata, A. Optimization of enzyme-mediated calcite precipitation as a soil-improvement technique: The effect of aragonite and gypsum on the mechanical properties of treated sand. Crystals 2017, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.K. Enzymes: Principles and biotechnological applications. Essays Biochem. 2015, 59, 1–41. [Google Scholar] [CrossRef]
- Segel, I. Enzyme Kinetics; John Wiley & Sons: New York, NY, USA, 1975. [Google Scholar]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Hanes, C.S. Studies on plant amylasesthe effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 1932, 26, 1406–1421. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J. Graphical methods in enzyme chemistry. Nature 1957, 179, 832. [Google Scholar] [CrossRef]
- Eadie, G.S. The inhibition of cholinesterase by physostigmine and prostigmine. J. Biol. Chem. 1942, 146, 85–93. [Google Scholar] [CrossRef]
- Liao, F.; Zhu, X.-Y.; Wang, Y.-M.; Zuo, Y.-P. The comparison of the estimation of enzyme kinetic parameters by fitting reaction curve to the integrated michaelis–menten rate equations of different predictor variables. J. Biochem. Bioph. Methods 2005, 62, 13–24. [Google Scholar] [CrossRef]
- Atkins, G.L.; Nimmo, I.A. The reliability of michaelis constants and maximum velocities estimated by using the integrated michaelis–menten equation. Biochem. J. 1973, 135, 779–784. [Google Scholar] [CrossRef]
- Orsi, B.A. Kinetic analysis of progress curves. Methods Enzymol. 1979, 63, 159–183. [Google Scholar] [PubMed]
- Newman, P.F.; Atkins, G.L.; Nimmo, I.A. The effect of systematic error on the accuracy of michaelis constants and maximum velocities estimated by using the integrated michaelis–menten equation. Biochem. J. 1974, 143, 779–781. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.A.; Goody, R.S. The original michaelis constant: Translation of the 1913 michaelis–menten paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef] [Green Version]
- Goličnik, M. The integrated michaelis-menten rate equation: Déjà vu or vu jàdé? J. Enzym. Inhib. Med. Chem. 2013, 28, 879–893. [Google Scholar] [CrossRef]
- Wen, K.J.; Li, Y.; Amini, F.; Li, L. Impact of bacteria and urease concentration on precipitation kinetics and crystal morphology of calcium carbonate. Acta Geotech. 2020, 15, 17–27. [Google Scholar] [CrossRef]
- Cesareo, S.D.; Langton, S.R. Kinetic properties of helicobacter pylori urease compared with jack bean urease. FEMS Microbiol. Lett. 1992, 99, 15–21. [Google Scholar] [CrossRef]
- Dixon, N.E.; Riddles, P.W.; Gazzola, C.; Blakeley, R.L.; Zerner, B. Jack bean urease (ec 3.5. 1.5). V. On the mechanism of action of urease on urea, formamide, acetamide, n-methylurea, and related compounds. Can. J. Biochem. 1980, 58, 1335–1344. [Google Scholar] [CrossRef]
- Tipton, K.F.; Dixon, H.B. Effects of ph on enzymes. Methods Enzymol. 1979, 63, 183–234. [Google Scholar] [PubMed]
- Fidaleo, M.; Lavecchia, R. Kinetic study of enzymatic urea hydrolysis in the ph range 4–9. Chem. Biochem. Eng. Q. 2003, 17, 311–318. [Google Scholar]
- Krajewska, B.; van Eldik, R.; Brindell, M. Temperature-and pressure-dependent stopped-flow kinetic studies of jack bean urease. Implications for the catalytic mechanism. J. Biol. Inorg. Chem. 2012, 17, 1123–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, A.; Michel, H. A contribution on the mechanism of the enzymatic cleavage of urea. Biochem. Physiol. Pflanzen 1972, 163, 103–109. [Google Scholar]
- Krajewska, B.; Ciurli, S. Jack bean (canavalia ensiformis) urease. Probing acid–base groups of the active site by ph variation. Plant Physiol. Biochem. 2005, 43, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.B.F.; Cruz, M.E.M.; Cabral, J.M.; Kennedy, J.F. Urease immobilization on an alkylamine derivative of titanium (iv)-porous silica: Kinetics and operational stability. J. Chem. Technol. Biotechnol. 1987, 39, 201–213. [Google Scholar] [CrossRef]
- Huang, T.C.; Chen, D.H. Kinetic study of urease-catalysed urea hydrolysis. J. Chem. Technol. Biotechnol. 1991, 52, 433–444. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N.; Hirata, A. Application of magnesium to improve uniform distribution of precipitated minerals in 1-m column specimens. Geomech. Eng. 2017, 12, 803–813. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N.; Neupane, D.; Lu, C. Effect of magnesium as substitute material in enzyme-mediated calcite precipitation for soil-improvement technique. Front. Bioeng. Biotechnol. 2016, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Jada, A.; Jradi, K. Role of polyelectrolytes in crystallogenesis of calcium carbonate. In Macromolecular Symposia; Wiley Online Library: Weinheim, Germany, 2006; pp. 147–151. [Google Scholar]
- Yashchenok, A.; Parakhonskiy, B.; Donatan, S.; Kohler, D.; Skirtach, A.; Möhwald, H. Polyelectrolyte multilayer microcapsules templated on spherical, elliptical and square calcium carbonate particles. J. Mater. Chem. B 2013, 1, 1223–1228. [Google Scholar] [CrossRef]
- Williams, F.V.; Ruehrwein, R.A. Effect of polyelectrolytes on the precipitation of calcium carbonate. J. Am. Chem. Soc. 1957, 79, 4898–4900. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N. Optimum condition for the application of enzyme-mediated calcite precipitation technique as soil improvement technique. Int. J. Adv. Sci. Eng. Inf. Technol. 2017, 7, 2145–2151. [Google Scholar] [CrossRef]
- Hoare, J.; Laidler, K. The molecular kinetics of the urea-urease system. Ii. The inhibition by products1. J. Am. Chem. Soc. 1950, 72, 2487–2489. [Google Scholar] [CrossRef]
- Leszko, M.; Zaborska, W.; Krajewska, B. Urease-catalyzed hydrolysis of urea differential vs. Integration kinetic methods. Bull. Pol. Acad. Sci.-Chem. 1997, 45, 129–138. [Google Scholar]
- Van Slyke, D.D.; Cullen, G.E. The mode of action of urease and of enzymes in general. J. Biol. Chem. 1914, 19, 141–180. [Google Scholar] [CrossRef]
- Schäfer, U.K.; Kaltwasser, H. Urease from staphylococcus saprophyticus: Purification, characterization and comparison to staphylococcus xylosus urease. Arch. Microbiol. 1994, 161, 393–399. [Google Scholar]
- Pham, V.P.; Nakano, A.; Van Der Star, W.R.; Heimovaara, T.J.; Van Paassen, L.A. Applying micp by denitrification in soils: A process analysis. Environ. Geotech. 2016, 5, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Rodriguez, A.; Quiroz-Limon, J.; Martins, A.M.; Peralta, H.; Avila-Calderon, E.; Sriranganathan, N.; Boyle, S.M.; Lopez-Merino, A. Enzymatic, immunological and phylogenetic characterization of brucella suis urease. BMC Microbiol. 2008, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Todd, M.J.; Hausinger, R. Competitive inhibitors of klebsiella aerogenes urease. Mechanisms of interaction with the nickel active site. J. Biol. Chem. 1989, 264, 15835–15842. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Ciurli, S.; Mangani, S. The complex of bacillus pasteurii urease with acetohydroxamate anion from x-ray data at 1.55 å resolution. JBIC J. Biol. Inorg. Chem. 2000, 5, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, B.; Brindell, M. Thermodynamic study of competitive inhibitors’ binding to urease. J. Therm. Anal. Calorim. 2016, 123, 2427–2439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Mulrooney, S.B.; Leung, A.F.; Zeng, Y.; Ko, B.B.; Hausinger, R.P.; Sun, H. Inhibition of urease by bismuth (iii): Implications for the mechanism of action of bismuth drugs. Biometals 2006, 19, 503–511. [Google Scholar] [CrossRef]
- Krajewska, B.; Zaborska, W. Double mode of inhibition-inducing interactions of 1, 4-naphthoquinone with urease: Arylation versus oxidation of enzyme thiols. Biorg. Med. Chem. 2007, 15, 4144–4151. [Google Scholar] [CrossRef]
- Zaborska, W.; Krajewska, B.; Kot, M.; Karcz, W. Quinone-induced inhibition of urease: Elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg. Chem. 2007, 35, 233–242. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Musiani, F.; Ciurli, S. Inactivation of urease by 1, 4-benzoquinone: Chemistry at the protein surface. Dalton Trans. 2016, 45, 5455–5459. [Google Scholar] [CrossRef] [Green Version]
- Kot, M.; Zaborska, W. Inhibition of jack bean urease by tetrachloro-o-benzoquinone and tetrachloro-p-benzoquinone. J. Enzym. Inhib. Med. Chem. 2006, 21, 537–542. [Google Scholar] [CrossRef]
- Krajewska, B.; Zaborska, W.a.; Leszko, M. Inhibition of chitosan-immobilized urease by slow-binding inhibitors: Ni2+, f− and acetohydroxamic acid. J. Mol. Catal. B Enzym. 2001, 14, 101–109. [Google Scholar] [CrossRef]
- Krajewska, B. Mono-(ag, hg) and di-(cu, hg) valent metal ions effects on the activity of jack bean urease. Probing the modes of metal binding to the enzyme. J. Enzym. Inhib. Med. Chem. 2008, 23, 535–542. [Google Scholar] [CrossRef]
- Moghal, A.A.B.; Lateef, M.A.; Mohammed, S.A.S.; Ahmad, M.; Usman, A.R.; Almajed, A. Heavy metal immobilization studies and enhancement in geotechnical properties of cohesive soils by eicp technique. Appl. Sci. 2020, 10, 7568. [Google Scholar] [CrossRef]
- Benini, S.; Cianci, M.; Mazzei, L.; Ciurli, S. Fluoride inhibition of sporosarcina pasteurii urease: Structure and thermodynamics. JBIC J. Biol. Inorg. Chem. 2014, 19, 1243–1261. [Google Scholar] [CrossRef]
- Krajewska, B.; Zaborska, W.; Leszko, M.; Brzózka, Z. Inhibition of jack bean urease by a mixture of boric acid and phosphate buffer ph 6.96. Pol. J. Chem. 1999, 73, 359–366. [Google Scholar]
- Reddy, M.S. Biomineralization of calcium carbonates and their engineered applications: A review. Front. Microbiol. 2013, 4, 314. [Google Scholar]
- McCarty, G.; Bremner, J.; Lee, J. Inhibition of plant and microbial ureases by phosphoroamides. Plant Soil 1990, 127, 269–283. [Google Scholar] [CrossRef]
- Dixon, N.E.; Hinds, J.A.; Fihelly, A.K.; Gazzola, C.; Winzor, D.J.; Blakeley, R.L.; Zerner, B. Jack bean urease (ec 3.5. 1.5). Iv. The molecular size and the mechanism of inhibition by hydroxamic acids. Spectrophotometric titration of enzymes with reversible inhibitors. Can. J. Biochem. 1980, 58, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, B. Urease immobilized on chitosan membrane. Inactivation by heavy metal ions. J. Chem. Technol. Biotechnol. 1991, 52, 157–162. [Google Scholar] [CrossRef]
- Yasuhara, H.; Neupane, D.; Hayashi, K.; Okamura, M. Experiments and predictions of physical properties of sand cemented by enzymatically-induced carbonate precipitation. Soils Found. 2012, 52, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Kavazanjian, E.; Hamdan, N. Enzyme induced carbonate precipitation (eicp) columns for ground improvement. In IFCEE 2015; American Society of Civil Engineers: Reston, VA, USA, 2015; pp. 2252–2261. [Google Scholar]
- Rahman, M.M.; Hora, R.N.; Ahenkorah, I.; Beecham, S.; Karim, M.R.; Iqbal, A. State-of-the-Art Review of Microbial-Induced Calcite Precipitation and Its Sustainability in Engineering Applications. Sustainability 2020, 12, 6281. [Google Scholar] [CrossRef]
- Dakhane, A.; Das, S.; Hansen, H.; O’Donnell, S.; Hanoon, F.; Rushton, A.; Perla, C.; Neithalath, N. Crack healing in cementitious mortars using enzyme-induced carbonate precipitation: Quantification based on fracture response. J. Mater. Civ. Eng. 2018, 30, 04018035. [Google Scholar] [CrossRef] [Green Version]
- Hamdan, N.; Kavazanjian, E., Jr.; O’Donnell, S. Carbonate Cementation via Plant Derived Urease. In Proceedings of the 18th International Conference on Soil Mechanics and Geotechnical Engineering, Paris, France, 2–6 September 2013. [Google Scholar]
- Dilrukshi, R.; Nakashima, K.; Kawasaki, S. Soil improvement using plant-derived urease-induced calcium carbonate precipitation. Soils Found. 2018, 58, 894–910. [Google Scholar] [CrossRef]
- Simatupang, M.; Okamura, M. Liquefaction resistance of sand remediated with carbonate precipitation at different degrees of saturation during curing. Soils Found. 2017, 57, 619–631. [Google Scholar] [CrossRef]
- Putra, H.; Yasuhara, H.; Kinoshita, N.; Fauzan, M. Promoting precipitation technique using bio-chemical grouting for soil liquefaction prevention. Civil Eng. Dimens. 2020, 22, 1–5. [Google Scholar] [CrossRef]
- Dilrukshia, R.; Kawasakib, S. Plant-derived urease induced sand cementation used in geotechnical engineering applications. In Proceedings of the International Conference on Geomechanics, Geo-Energy and Geo-Resources, Melbourne, Australia, 28–29 September 2016. [Google Scholar]
- Khodadadi, T.H.; Javadi, N.; Krishnan, V.; Hamdan, N.; Kavazanjian, E.J. Crude urease extract for biocementation. J. Mater. Civil Eng. 2020, 32, 04020374. [Google Scholar] [CrossRef]
- Knorr, B. Enzyme-Induced Carbonate Precipitation for the Mitigation of Fugitive Dust. Ph.D. Thesis, Arizona State University, Tempe, AZ, USA, 2014. [Google Scholar]
- Cuccurullo, A.; Gallipoli, D.; Bruno, A.W.; Augarde, C.; Hughes, P.; La Borderie, C. Soil stabilization against water erosion via calcite precipitation by plant-derived urease. In Proceedings of the National Conference of the Researchers of Geotechnical Engineering, Lecco, Italy, 3–5 July 2019; pp. 753–762. [Google Scholar]
- Bang, S.C.; Min, S.H.; Bang, S.S. Kgs awards lectures: Application of microbiologically induced soil stabilization technique for dust suppression. Int. J. Geo-Eng. 2011, 3, 27–37. [Google Scholar]
- Bang, S.S.; Bang, S.; Frutiger, S.; Nehl, L.M.; Comes, B.L. Application of novel biological technique in dust suppression. In Proceedings of the 88th Transportation Research Board Annual Meeting, Washington, DC, USA, 11–15 January 2009. [Google Scholar]
- Lo, C.-Y.; Tirkolaei, H.K.; Hua, M.; De Rosa, I.M.; Carlson, L.; Kavazanjian, E., Jr.; He, X. Durable and ductile double-network material for dust control. Geoderma 2020, 361, 114090. [Google Scholar] [CrossRef]
- Woolley, M.A.; Van Paassen, L.; Kavazanjian, E., Jr. Impact on surface hydraulic conductivity of eicp treatment for fugitive dust mitigation. In Geo-Congress 2020: Biogeotechnics; American Society of Civil Engineers: Reston, VA, USA, 2020; pp. 132–140. [Google Scholar]
- Almajed, A.; Lemboye, K.; Arab, M.G.; Alnuaim, A. Mitigating wind erosion of sand using biopolymer-assisted eicp technique. Soils Found. 2020, 60, 356–371. [Google Scholar] [CrossRef]
- Liu, K.-W.; Jiang, N.-J.; Qin, J.-D.; Wang, Y.-J.; Tang, C.-S.; Han, X.-L. An experimental study of mitigating coastal sand dune erosion by microbial-and enzymatic-induced carbonate precipitation. Acta Geotech. 2021, 16, 467–480. [Google Scholar] [CrossRef]
- Miao, L.; Wu, L.; Sun, X. Enzyme-catalysed mineralisation experiment study to solidify desert sands. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ossai, R.; Rivera, L.; Bandini, P. Experimental study to determine an eicp application method feasible for field treatment for soil erosion control. In Geo-Congress 2020: Biogeotechnics; American Society of Civil Engineers: Reston, VA, USA, 2020; pp. 205–213. [Google Scholar]
- Nemati, M.; Greene, E.; Voordouw, G. Permeability profile modification using bacterially formed calcium carbonate: Comparison with enzymic option. Process Biochem. 2005, 40, 925–933. [Google Scholar] [CrossRef]
- Nemati, M.; Voordouw, G. Modification of porous media permeability, using calcium carbonate produced enzymatically in situ. Enzym. Microb. Technol. 2003, 33, 635–642. [Google Scholar] [CrossRef]
- Song, J.Y.; Sim, Y.; Jang, J.; Hong, W.T.; Yun, T.S. Near-surface soil stabilization by enzyme-induced carbonate precipitation for fugitive dust suppression. Acta Geotech. 2020, 15, 1967–1980. [Google Scholar] [CrossRef]
- Hamdan, N.; Kavazanjian, E. Enzyme-induced carbonate mineral precipitation for fugitive dust control. Geotechnique 2016, 66, 546–555. [Google Scholar] [CrossRef]
- Nam, I.-H.; Roh, S.-B.; Park, M.-J.; Chon, C.-M.; Kim, J.-G.; Jeong, S.-W.; Song, H.; Yoon, M.-H. Immobilization of heavy metal contaminated mine wastes using canavalia ensiformis extract. Catena 2015, 136, 53–58. [Google Scholar] [CrossRef]
- Moghal, B.A.A.; Lateef, M.A.; Mohammed, S.A.S.; Lemboye, K.K.; Chittoori, B.C.S.; Almajed, A. Efficacy of enzymatically induced calcium carbonate precipitation in the retention of heavy metal ions. Sustainability 2020, 12, 7019. [Google Scholar] [CrossRef]












| Method | Advantage | Limitation |
|---|---|---|
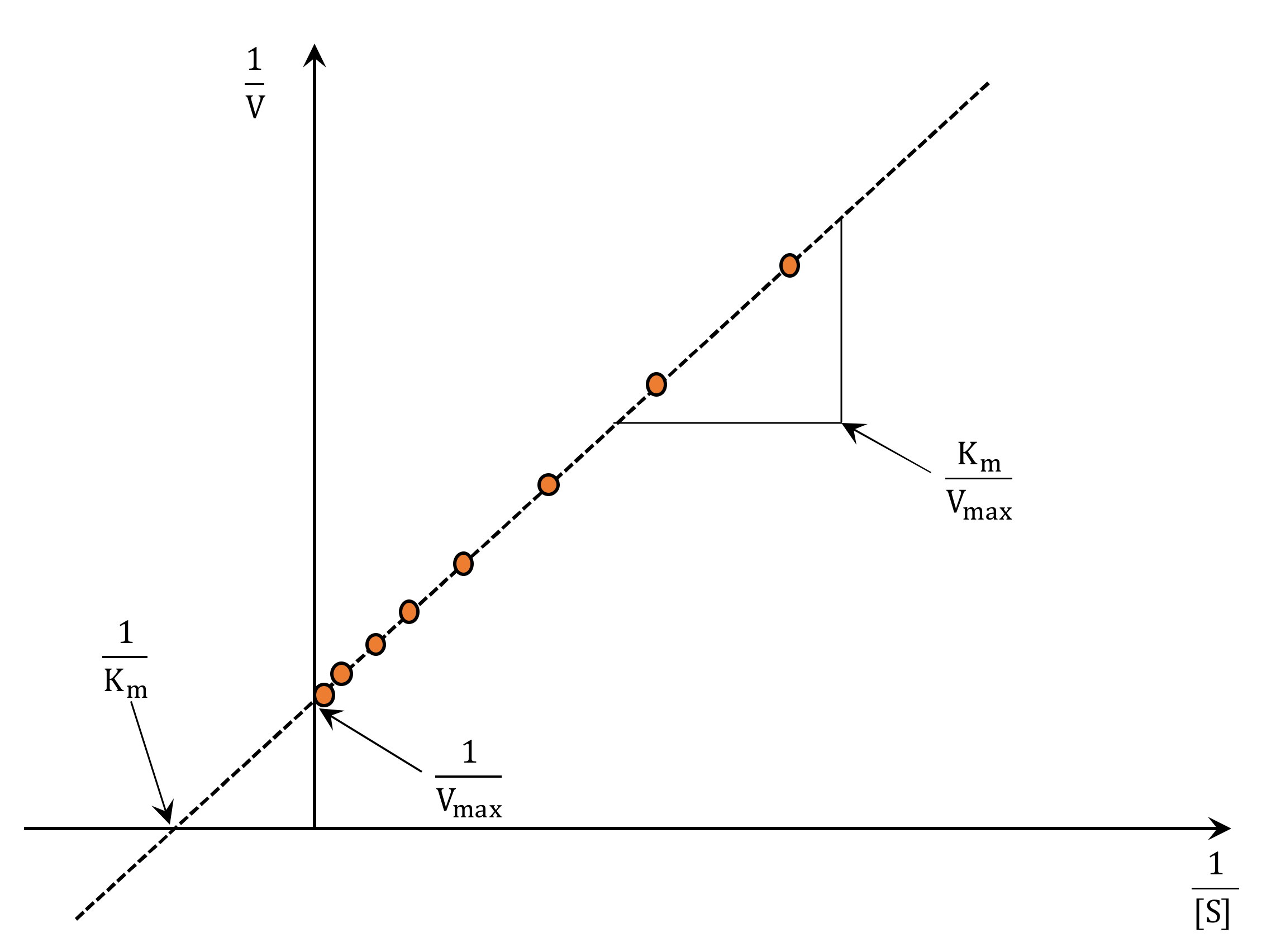
| Lineweaver and Burk [57] | (1) Gives a good estimation of and . | (1) Poor fit between data and straight line. (2) Large error at low where measurements are less accurate. |
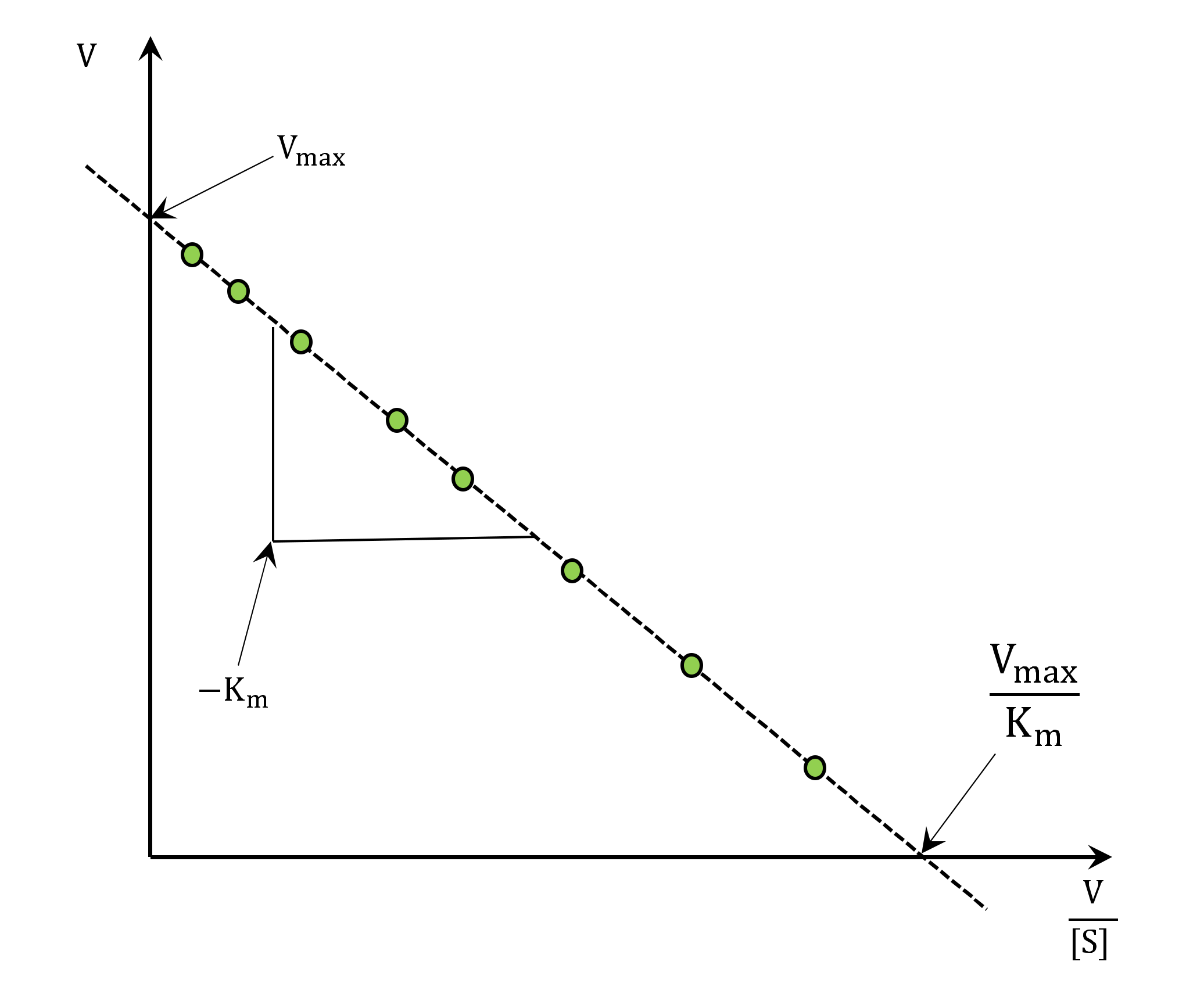
| Hanes–Woolf plot [58] | (1) Direct determination of and . | (1) Both axes contain an independent variable. (2) large error at low . |
| Eadie-Hofstee plot [60] | (1) Less error observed at low . | (1) Data are usually scattered. (2) The dependent variable (V) occurs in both the X- and Y-axes. |
| Kinetic Parameters | Value | Unit | Reference |
|---|---|---|---|
| 18.3 ± 0.05 | min−1 | Fidaleo and Lavecchia [71] | |
| 3.21 ± 0.36 | mmol/L | Fidaleo and Lavecchia [71] | |
| 12.2 ± 0.11 | mmol/L | Fidaleo and Lavecchia [71] | |
| 7.57 ± 0.41 × 10−4 | mmol/L | Fidaleo and Lavecchia [71] | |
| 1.27 ± 0.08 × 10−5 | mmol/L | Fidaleo and Lavecchia [71] | |
| 32.6–35.8 | kJ/mol | Martins, Cruz [75], Huang and Chen [76], Fidaleo and Lavecchia [71] | |
| 416.6 ± 0.50 | K | Fidaleo and Lavecchia [71] |
| Inhibitors | Type of Inhibition | Reference |
|---|---|---|
| Ammonium ion/ammonium carbonate (product) | Non-competitive | [83,84,85] |
| Urea analogues (e.g., Hydroxyurea, Formamide, Thiourea, Ethylurea and Methylurea) | Competitive | [86,87,88] |
| Thiols (e.g., β-Mercaptoethanol) | Competitive | [89,90,91] |
| Bismuth compounds | Competitive | [92] |
| Quinones (e.g., 1,4-Benzoquinone, 2,5-Dimethyl-1,4-benzoquinone and Tetrachloro-1,4-benzoquinone) | Competitive (slow binding) | [93,94,95,96] |
| Hydroxamic acids | [34,89,90,97] | |
| Heavy metal ions (e.g., Hg2+, Ag+, Cu2+, Mg2+, Zn2+, Cd2+, Ni2+, Pb2+ and Co2+) | Competitive (slow binding) | [78,94,98,99] |
| Fluoride | Uncompetitive (slow binding) | [34,100] |
| Boron compounds (e.g., Boric acid, Butylboronic acid, Phenylboronic acid and 4-Bromophenylboronic acid) | Competitive | [34,91,101,102] |
| Phosphate buffer | Competitive | [101] |
| Amides and esters of phosphoric acid (e.g., Phosphoric triamide (PTA), Phenylphosphorodiamidate (PPD), 4-Chlorophenylphosphorodiamidate, N-(diaminophosphinyl)-benzamide and N-(diaminophoshinyl)-4-fluoro-benzamide) | Competitive (slow binding) | [14,34,89,103] |
| Acylhydroxamic acids (e.g., Acetohydroxamic acid) | Competitive (slow binding) | [97,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahenkorah, I.; Rahman, M.M.; Karim, M.R.; Beecham, S.; Saint, C. A Review of Enzyme Induced Carbonate Precipitation (EICP): The Role of Enzyme Kinetics. Sustain. Chem. 2021, 2, 92-114. https://doi.org/10.3390/suschem2010007
Ahenkorah I, Rahman MM, Karim MR, Beecham S, Saint C. A Review of Enzyme Induced Carbonate Precipitation (EICP): The Role of Enzyme Kinetics. Sustainable Chemistry. 2021; 2(1):92-114. https://doi.org/10.3390/suschem2010007
Chicago/Turabian StyleAhenkorah, Isaac, Md Mizanur Rahman, Md Rajibul Karim, Simon Beecham, and Christopher Saint. 2021. "A Review of Enzyme Induced Carbonate Precipitation (EICP): The Role of Enzyme Kinetics" Sustainable Chemistry 2, no. 1: 92-114. https://doi.org/10.3390/suschem2010007
APA StyleAhenkorah, I., Rahman, M. M., Karim, M. R., Beecham, S., & Saint, C. (2021). A Review of Enzyme Induced Carbonate Precipitation (EICP): The Role of Enzyme Kinetics. Sustainable Chemistry, 2(1), 92-114. https://doi.org/10.3390/suschem2010007