1. Introduction
Electrochemical polishing or electropolishing is the controlled corrosion of metal surfaces [
1]. The concept behind this mechanism of liquids corroding metals is to reduce the surface roughness of the polished metals [
2,
3]. Unlike surface buffing alternatives, electropolishing can reduce surface roughness and impurities to nearly negligible quantities on polished surfaces. Currently, large quantities of surface-polished products are being treated with hazardous chemical solutions, such as phosphoric and sulfuric acids that are used to electropolish stainless steel [
4].
Recently, the benefits of electrochemical polishing are gaining more recognition as an ideal method for improving a metal’s optimum roughness that can also greatly improve electrical conductivity [
5]. Many acid treatments currently utilized for electropolishing metal surfaces provide an ideal mirror finish by removing the exposed surface layer of the sheet metal. However, acid solutions provide this clean, electropolished finish to the metal at the expense of hydrogen contamination [
2,
5]. The removal of hydrogen contamination generally entails the use of high temperature treatments in excess of 800 °C for several hours in vacuo [
6].
Deep eutectic solvents have shown a great promise for many electrochemical applications [
1,
2,
3,
4,
5,
6,
7,
8]. The main advantages for deep eutectic solvents over state-of-the-art acidic electropolishing are the wide potential windows and high conductivity compared to other non-aqueous solvents [
1,
2,
3,
4,
5,
6,
7,
8].
In the current study, high-purity metal samples of copper will be tested to determine the effectiveness of both an industry standard acid solution (1 M phosphoric acid), and a deep eutectic solvent medium comprised of ethylene glycol and choline chloride mixed in a 2:1 molar ratio, respectively [
7,
8]. Several scientists have demonstrated the effectiveness of various conformities of this deep eutectic solvent with various metals and alloys [
1,
3,
9,
10]. It is the aim of this investigation to optimize the electrochemistry tests to produce a superior polish for copper metals, and to weigh the benefits of this deep eutectic solvent against an industry standard 1M phosphoric acid electropolishing treatment.
2. Materials and Methods
Materials: A 1 M phosphoric acid mixture was used for the acid treatments (Sigma-Aldrich, St. Louis, MO, USA, 85%). Acid-free treatments were carried out using a deep eutectic solvent comprised of choline chloride (Acros Organics, Fair Lawn, NJ, USA, 99%) and ethylene glycol (Sigma-Aldrich, St. Louis, MO, USA, 99.8%); both chemicals were used as received. The deep eutectic solvent mixture was created by stirring the two components together at a component ratio of 2 ethylene glycol: 1 choline chloride at 70 °C until a homogeneous colorless liquid emerged.
Samples Preparation: Samples of high-purity copper (>99.95%) were bored from supplied sheets (3 mm thickness) and labeled for use. In each test, five samples of each respective sample were taped with polyimide film tape to restrict electrochemical activity, but leaving (not covering with the tape) a 1 cm2 region on both the front and back faces of each metal to polish. This resulted in a 2 cm2 region of polishing for each sample when calculating current density. Metal samples were then immersed in the electropolishing solution of choice, such that the regions for polishing were completely immersed. Each metal was submitted to a cursory linear sweep voltammetry test to determine optimum voltage conditions to set for running chronoamperometry over a 900 s polishing sequence.
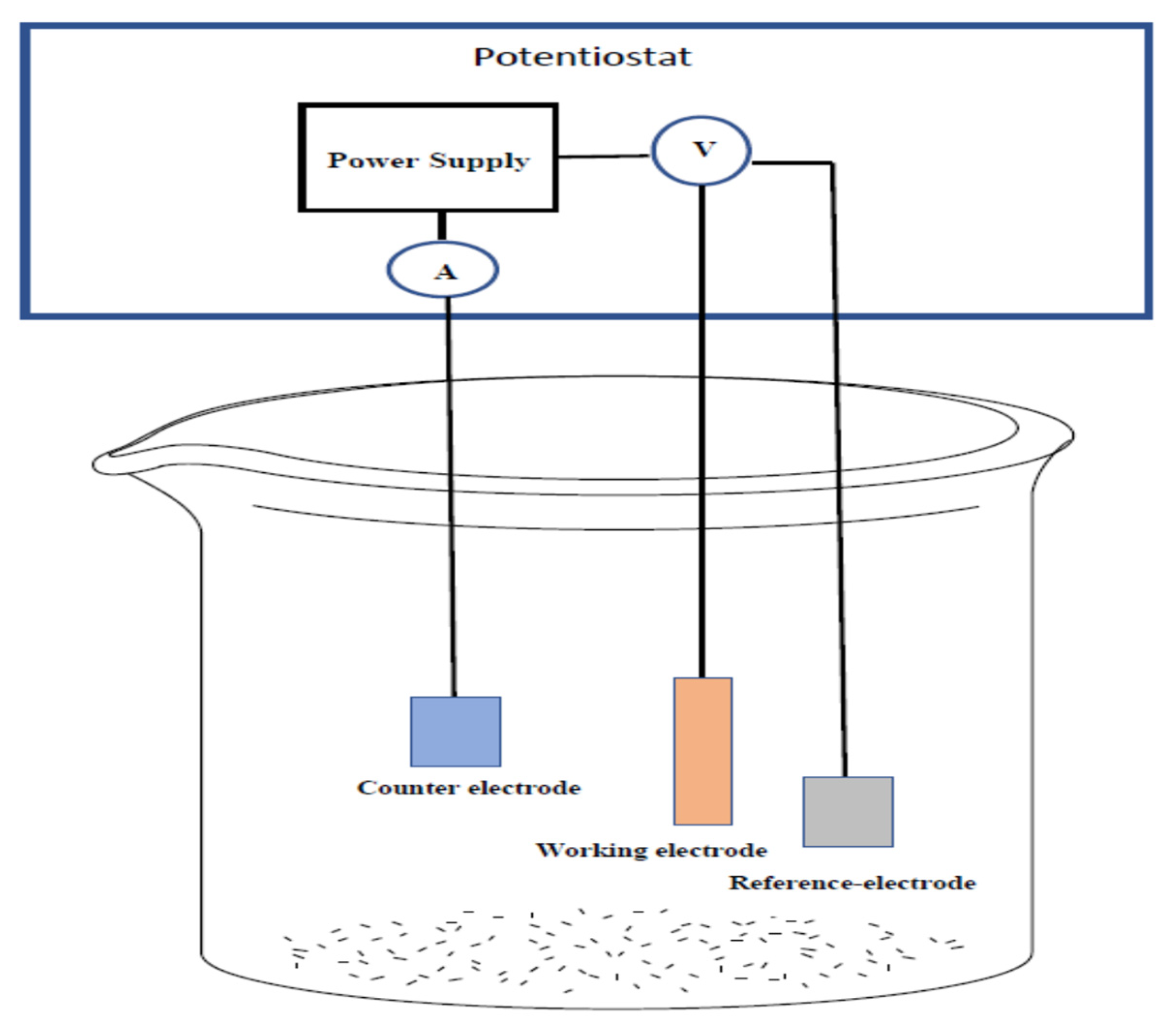
Electrochemical Setup and Measurements: The deep eutectic solvent’s effects on electropolishing the metal surfaces of interest were analyzed using the necessary machines. Voltammetry and chronoamperometry tests were conducted using a three electrodes system (
Scheme 1) connected to a Gamry PCI4-G750 potentiostat and controlled using the accompanying framework and
e-chem Analyst (v. 5.5) software.
The electropolishing procedure made use of a flat platinum counter electrode plate with a silver wire reference with a spacing of 2.5 cm between each electrode for the experimental setup. The temperature was maintained at 70 °C and flow control inside the beaker was induced by a slow rotation at 60 rpm using a 1-inch magnetic stir rod via a hotplate. Both electrodes were degreased using deionized water and acetone to preserve the purity of samples during testing. The working electrode was abraded with 150 grit glass paper, rinsed and dried prior to each recorded measurement to ensure reproducible voltammetric effects. Electrochemical measurements were performed at 70 °C with a constant scan rate of 20 mV/s used in voltammetric experiments.
Characterizations: An AFM surface analysis was carried out using a Dimension 3100 Nanoscope IV scanning probe microscope, manufactured by Digital Instruments, with software version 6.12 in tapping mode. Step height measurements were recorded in µm via the Alpha Step 200 by Tencor Instruments. A KH-1300 HIROX digital microscope was utilized for optical comparison to produce representative images scaled to 1600 × 1200 pixels. After completion of each experiment, samples were dried and weight was recorded using a high precision digital scale for calculations of mass loss due to electrochemical etching.
3. Results
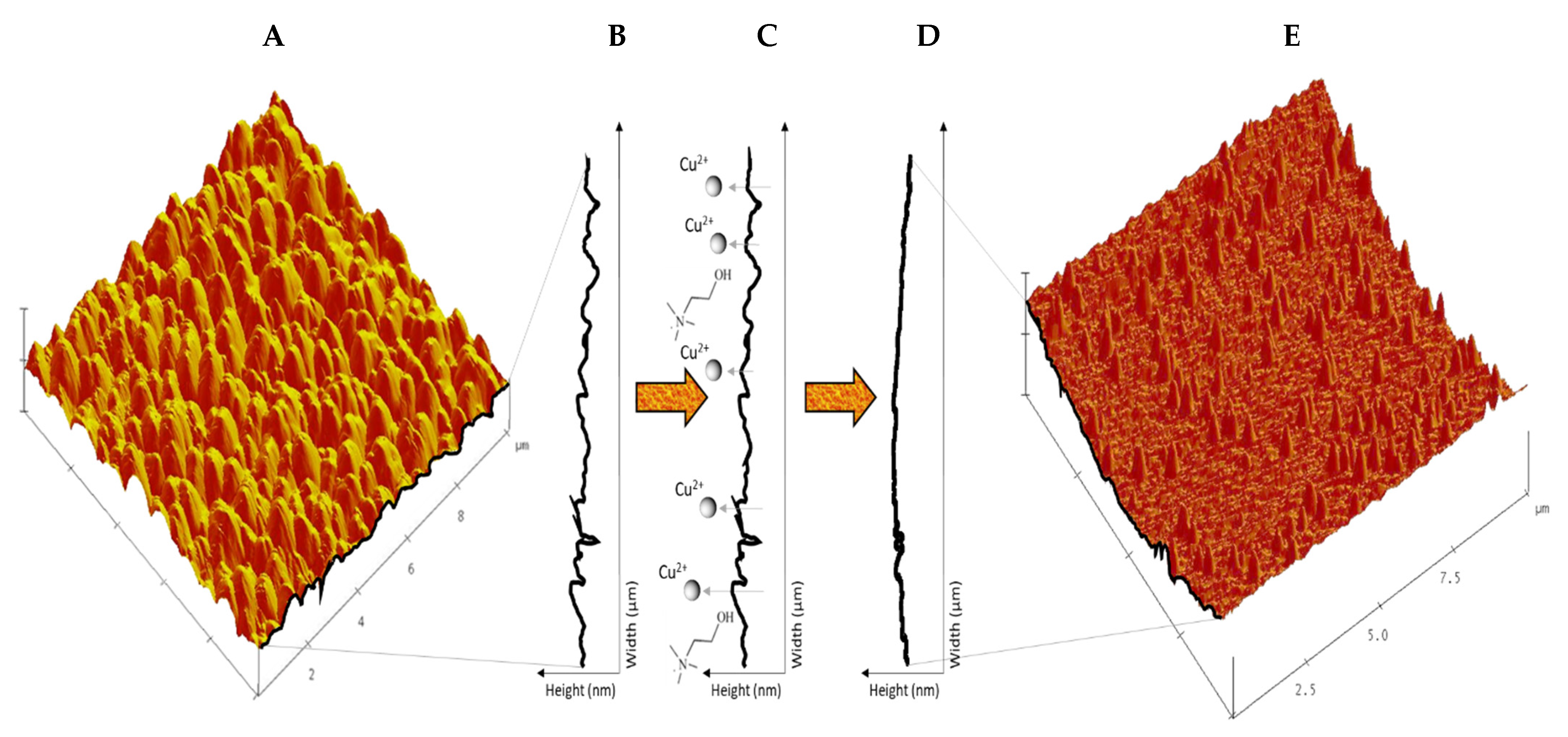
The mechanism for electropolishing copper metal surfaces using the deep eutectic solvent electrolyte is represented in
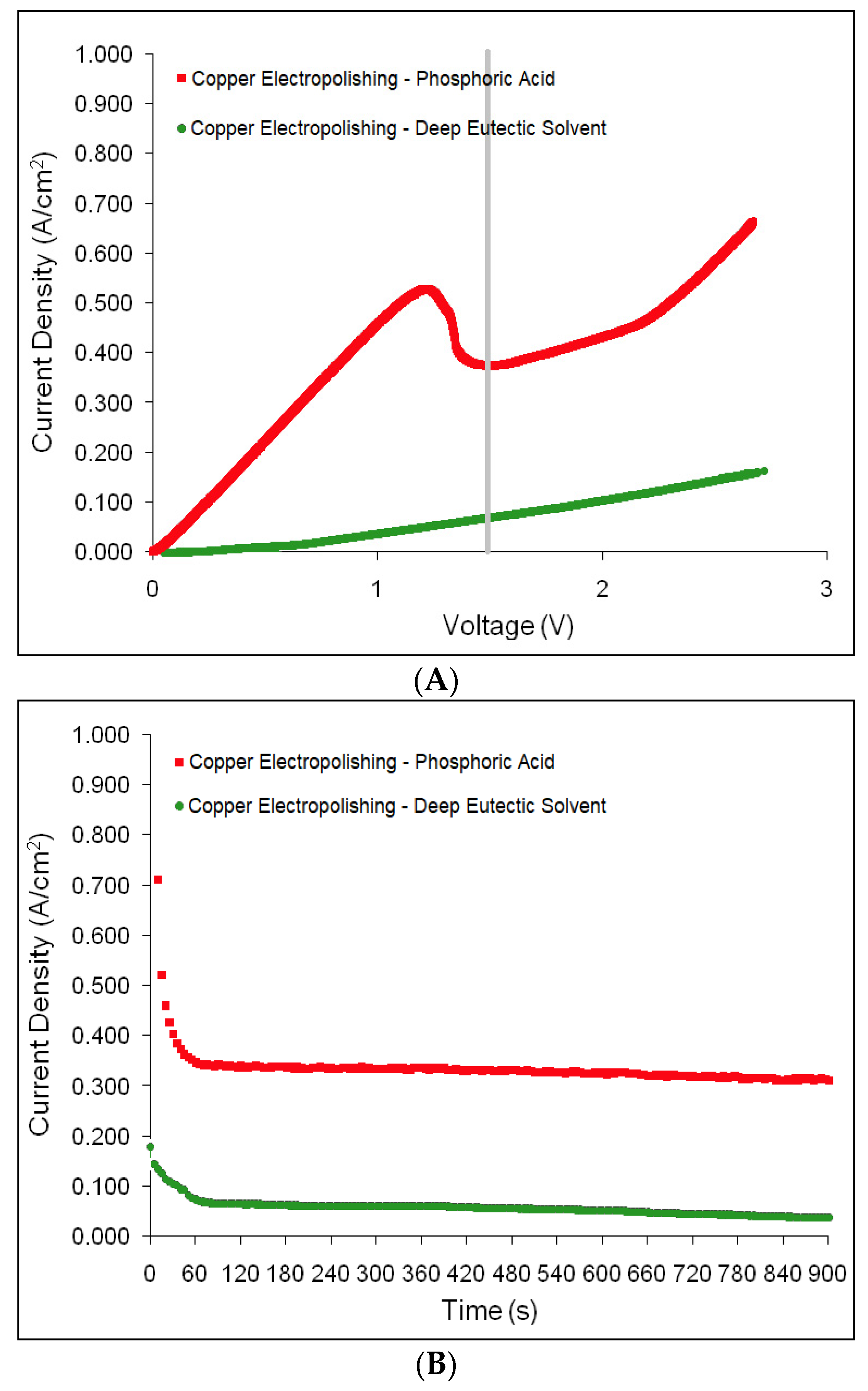
Figure 1. Upon comparison of voltammograms generated for both the deep eutectic solvent and phosphoric acid treatments of copper samples when stepped from 0 to 3 V at a constant scan rate of 20 mVs
−1, the ideal scenario for chronoamperometry settings for both electrolytes revealed to be 1.5 V (
Figure 2A). With a fixed voltage of 1.5 V, both chronoamperograms revealed electropolishing rates consistent with the currents reported from the I-V curve from linear sweep voltammetry with expressed means of 0.38 A/cm
2 and 0.06 A/cm
2 for the phosphoric acid and acid-free electrolytes, respectively (
Figure 2B). Having selected a fixed voltage of 1.5 V for electropolishing copper with the deep eutectic solvent, a reported current range of 0.04 to 0.08 A/cm
2 for the majority of chronoamperometry displayed a very slow rate of current decline (
Figure 2B). A comparison of mass before (7.382 ± 0.03 g) and mass post-electropolishing (7.329 ± 0.04 g) revealed a difference of 0.053 g with an electropolishing rate of 59.44 µg/s (
Table 1).
As-received copper samples returned an average roughness of 167 ± 9 nm upon scanning with AFM in tapping mode (
Table 2). When compared with a 14.7 ± 2 nm roughness average for post-treatment with the deep eutectic solvent medium (
Figure 3), a roughness comparison calculated a difference of 152.4 ± 3 nm (
Table 2). This resulted in a post-treatment surface 8× smoother than the original metal surface prior to electropolishing treatments (
Table 2). This differentiation in roughness accounts for a high percentage of smoothing efficiency, 91.1 ± 1.5% (
Table 2). This efficiency calculation provides the average roughness difference divided by the roughness prior to treatment (expressed as a percentage) permitted for the exposed 2 cm
2 region of the working electrode. Since this percentage cannot ever truly be zero, given that a smoothness of zero would inevitably be a frictionless surface, a value of 91.1 ± 1.5% is extremely high (
Table 2). The step height recorded for the deep eutectic solvent electropolishing treatment was 20.2 µm (
Table 3).
Electropolishing copper specimens with phosphoric acid revealed a steady incline from 0 to 1.25 V with an initial peak at 0.54 A/cm
2 and with an immediate decline to a local minimum of 0.37 A/cm
2 at 1.5 V. After this local minimum, the incline rate increased at 2.25 V and remained constant until nearly 3 V where, much like the deep eutectic solvent curve for copper electropolishing, it reached a point of irreproducibility (
Figure 2A).
After surveying the local maxima and minima present in the linear sweep voltammogram, 1.5 V was selected to run chronoamperometry (
Figure 2B). With the voltage fixed at 1.5 V, the current would not only be checked to authenticate the linear sweep voltammogram for relative accuracy, but the optimum voltage could be confirmed while revealing a resulting current density across the exposed surface used for polishing. The current range from chronoamperometry was 0.34 to 0.30 A/cm
2 in the form of a very gradual decline nearly identical to the chronoamperogram produced by the deep eutectic solvent, with the exception of a significantly higher current (
Figure 2B). When compared to the copper sample weight prior to electropolishing treatments, the mass difference was calculated to be 0.053 g for an electropolishing rate of 73.44 µg/s (
Table 1). Comparisons with other similar pure metals revealed that the smoothing efficiency of the deep eutectic solvent in this paper was greater than those observed in the external literature using the same deep eutectic solvent under similar electropolishing conditions (
Table 2).
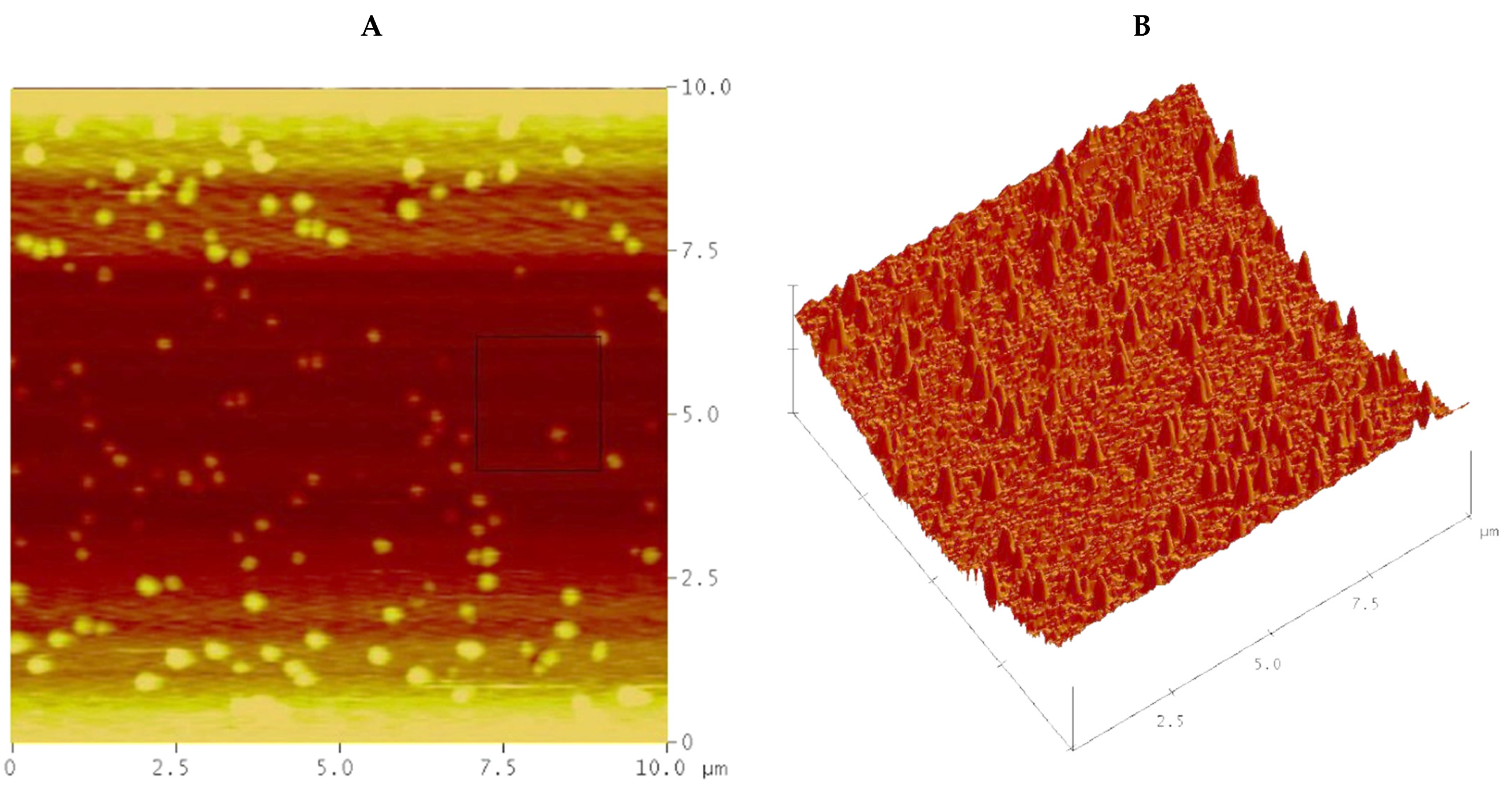
A recorded roughness average of 82.6 ± 6 nm for phosphoric acid electropolishing revealed that a roughness difference of 85.0 ± 5 nm exists between the original as-received sample roughness and the subsequent electrochemical testing reported (
Table 3). It can be noted that, although pitting affected the phosphoric acid representative polish of the total region, there were extreme peaks and troughs left due to pitting at low current densities. Hydrogen contamination can be observed to affect the overall roughness average of the region, with an average roughness of 16 ± 2 nm in a 4.5 × 4.5 µm recording region (
Figure 4A). A step height analysis revealed a vertical step distance of 24.0 µm etched away from the original copper metal surface during electropolishing (
Table 3).
4. Discussion
The electropolishing rate for copper with phosphoric acid was reported as 73.44 µg/s, whereas treatments via the deep eutectic solvent electrolyte yielded a less comprehensive polishing rate of 59.44 µg/s (
Table 1). When comparing step height of both samples, it was again confirmed that the etching efficiency was higher for phosphoric acid treatments with copper, measuring at 24.0 µm, compared to a step height of only 20.0 µm reported for deep eutectic solvent electropolishing results (
Table 3).
Roughness averages for copper samples using both electrolytes shared little in common (
Table 2). The roughness difference for the deep eutectic solvent was reported as 152.4 ± 3 nm, whereas phosphoric acid treatments reported an overall change in roughness from pre-treatment to post-treatment of 85.0 ± 5 nm (
Table 2). This difference being nearly double for the deep eutectic solvent mixture, the indication is clearly that the phosphoric acid mixtures are utilized for their superior rate of etching, and not for their overall smoothing efficiency [
9]. Providing roughness averages to consider, a post-treatment roughness analysis revealed heavy splotching in the copper samples treated with phosphoric acid, which is only further accentuated by a view of the accompanying 3D microscale terrain map. With a reported roughness average of 82.6 ± 6 nm, the phosphoric acid solution revealed significant peaks and residual splotches likely caused by oxidation at the metal’s surface during electropolishing (
Figure 4) [
10].
Throughout the 900-s electropolishing experiment, a chemical reaction occurred at the anode immersed in the deep eutectic solvent due to the oxidation of Cu and the formation of CuCl
2 (Equation (1)). This is notably similar to the literature documenting reactions with high-purity nickel noted in [
2]:
We observed the presence of trimethylamine, ethanol, ethylene glycol and other products, with the incidence of trimethylamine being accounted for by the Hofmann elimination of the choline base as choline hydroxide (Equation (2)):
The reduction reaction at the cathode (counter electrode) involves the decomposition of choline by the formation of a choline radical via the acceptance of an electron:
Thus, the transient choline radical, depicted in parentheses (Equation (3)), results from the addition of an electron from the anode at the cathode, and quickly decomposes to trimethylamine [
2]. A review of the literature also indicates that the residual pitting on the surface of the metal not only affects surface reflectivity, but also is likely to affect conductivity due to increased surface area [
11,
12,
13]. The plentiful abrasions and bumps generated during the electropolishing procedure (clearly visible in AFM imagery) are a result of bubbles formed on the metal’s surface during electropolishing. With significant proportions of the metal surface being deteriorated per second, the bubbling that occurs at the cathode can often leave these marks as they pop on the metal’s surface, sometimes marring the newly treated surface (
Figure 5B) [
12,
14].
Once electropolishing is complete, surface scrapes seem mostly absent from both the phosphoric acid and acid-free treatments. Although some of the surface reflectivity of the metal seems contained in the smoothed outlines of the present scratches, the roughness is relatively unaffected with an average of 14.7 ± 2 nm (
Figure 3) [
11]. With the aberrant marring of the copper sample during phosphoric acid electropolishing treatments, the roughness average greatly suffers (82.6 ± 6 nm), with intermittent peaks that caused the silicon tip attached to the AFM cantilever to become stuck during the trace and retrace of scattered spots on the copper metal surface. This occurred during two discarded post-electropolishing surveys, which required tip replacement and AFM recalibration (
Figure 4) [
2,
12].
This exchange is heavily recorded in the literature regarding electropolishing and extensive use of hydrogen evolution at the cathode as a driver for this type of pitting. The bubbles tend to form at points in the range when low current densities occur in the electropolishing procedure [
15]. This tends to happen towards the end of the trial, when most of the originally protruding surfaces to be polished and deteriorated away have been removed [
16,
17]. When this occurs, the associated chronoamperogram reports a slow and steady decline, as the remaining surface area available to be electropolished slowly decreases at the rate recorded in
Table 1.
Pitting tends to occur at low current densities, or when the current applied through the 2 cm
2 exposed surface of the metal is occurring over less and less surface area as the sample is being polished [
1,
18]. This pitting is relatively inevitable, since the concept of electropolishing is to always have less surface area than prior to treatment [
19]. With good fortune, this decreased surface area will be accompanied by the benefit of an overall smoother surface and heightened electroconductivity properties as electrical impulses will have less distance to dissipate along the microscale surface of the metal over significant distances [
13]. For this to occur, the protruding peaks,
Figure 1A,B, must somehow be electropolished at a faster rate than the average surface in the working electrode surface. Provided that all of these interactions occur appropriately at the metal’s surface and at the proper prescribed rates, an efficient electropolishing treatment can be achieved [
11].











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}