
Sialic Acid Ameliorates Cognitive Deficits by Reducing Amyloid Deposition, Nerve Fiber Production, and Neuronal Apoptosis in a Mice Model of Alzheimer’s Disease

 ,
,  , ,
, ,
Abstract
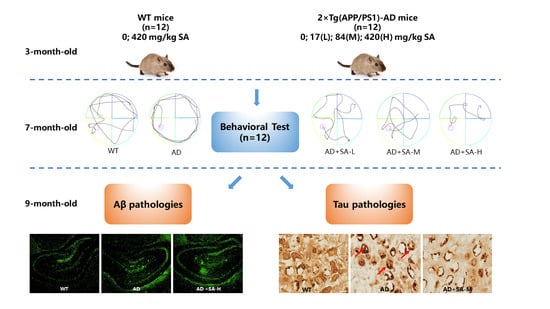
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Treatment
2.3. Behavioral Test
2.3.1. Morris Water Maze
2.3.2. Open-Field Test
2.4. Tissue and Serum Preparation
2.5. Serum Biochemical Analysis
2.6. Thioflavin T (ThT) Staining
2.7. Silver Staining
2.8. Nissl Staining
2.9. Statistical Analysis
3. Results
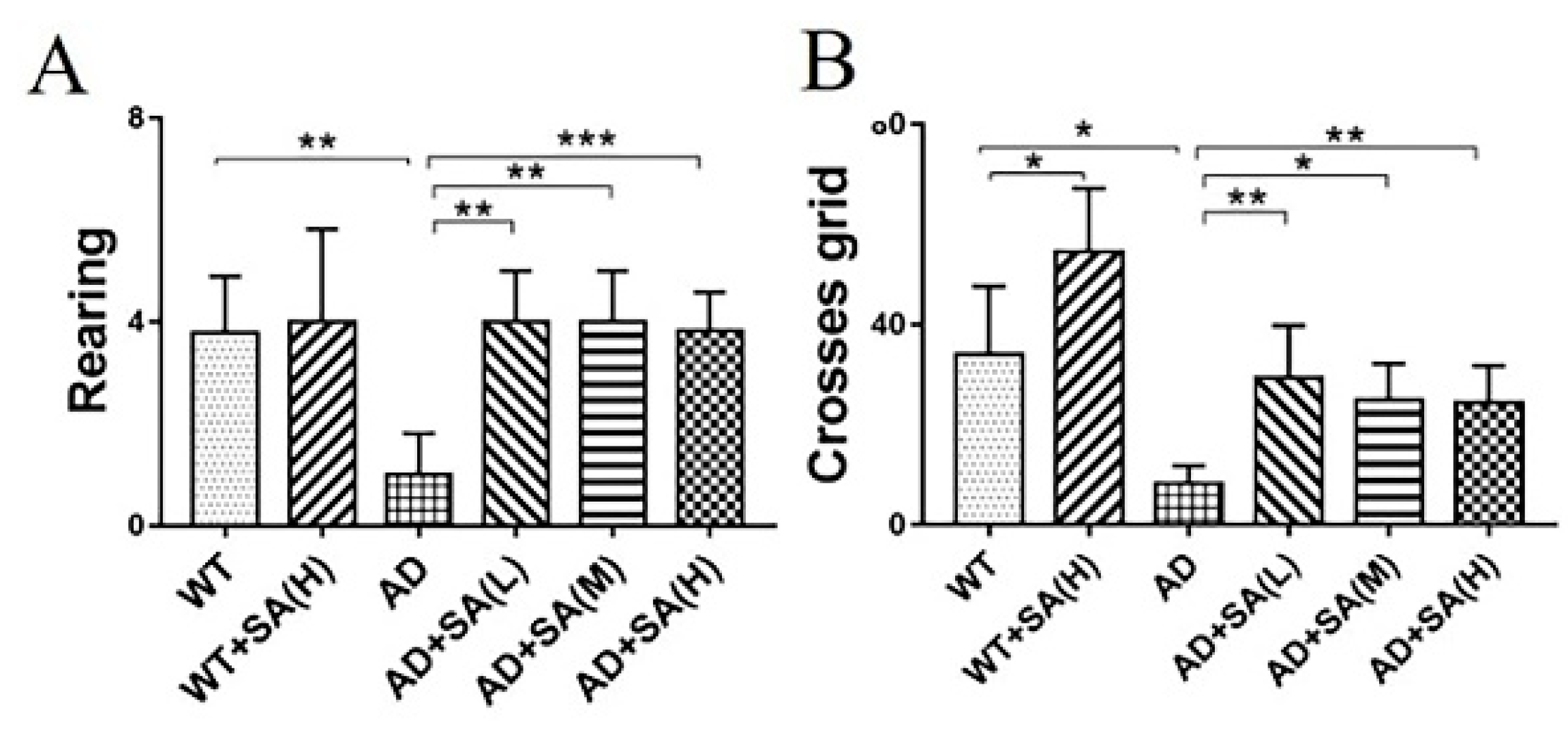
3.1. SA Treatment Ameliorated the Cognitive Impairment and Depression/Anxiety Behaviors in AD Mice
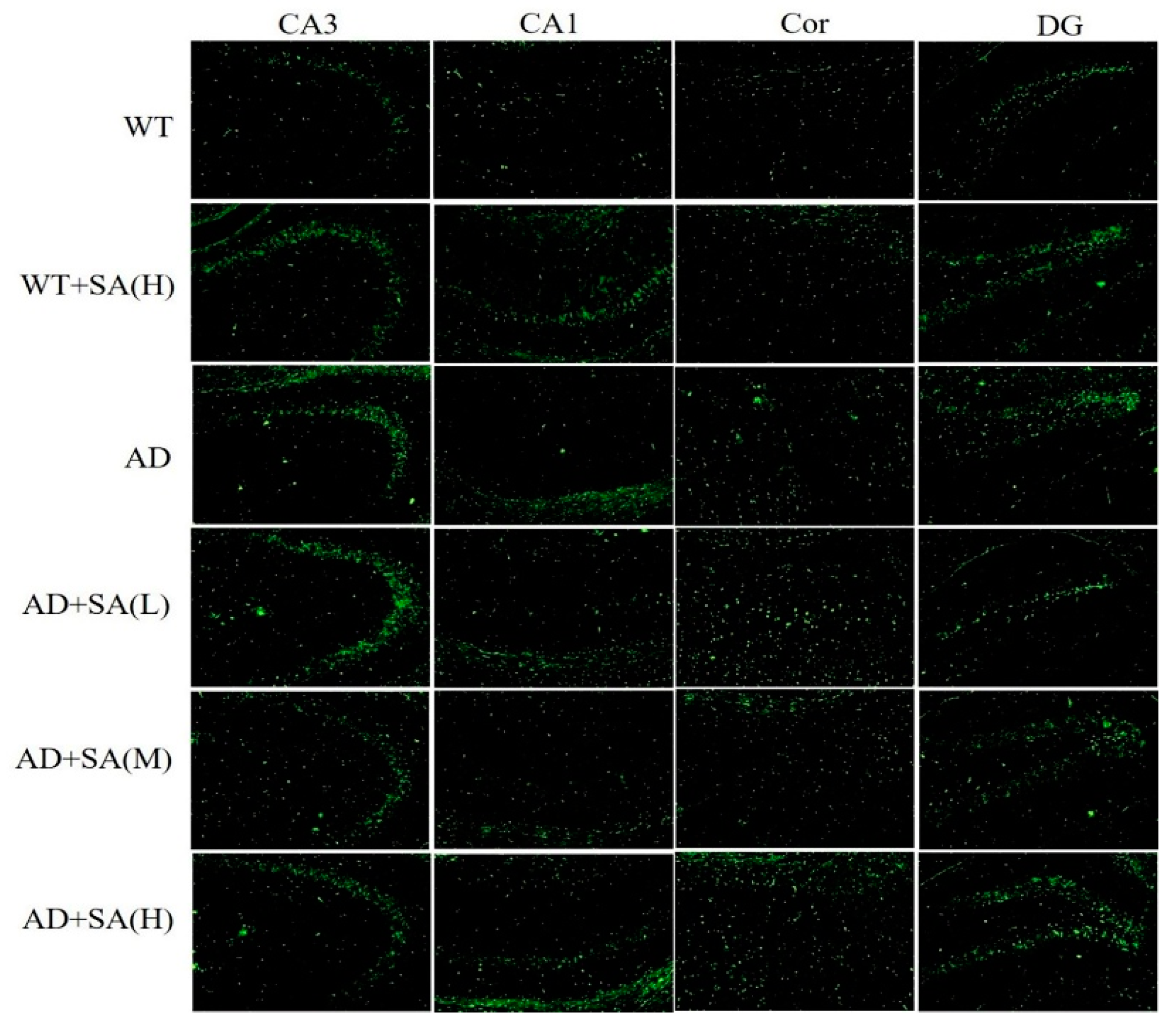
3.2. SA Significantly Suppressed the Pathological Hallmarks of AD Mice
3.3. SA Reversed Neuronal Loss in AD Mice in a Concentration-Independent Manner
3.4. SA Lowered Blood Lipids and May Prevent Vascular Diseases
3.5. SA May Affect Liver Function in AD Mice
3.6. SA Has a Little Effect on Inositol, Glycine and Taurine
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Penke, B.; Bogár, F.; Paragi, G.; Gera, J.; Fülöp, L. Key Peptides and Proteins in Alzheimer’s Disease. Curr. Protein Pept. Sci. 2019, 20, 577–599. [Google Scholar] [CrossRef]
- Chen, X.; Varki, A. Advances in the Biology and Chemistry of Sialic Acids. ACS Chem. Biol. 2009, 5, 163–176. [Google Scholar] [CrossRef]
- Schauer, R.; Kamerling, J.P. Exploration of the Sialic Acid World. Adv. Carbohydr. Chem. Biochem. 2018, 75, 1–213. [Google Scholar] [CrossRef]
- Wang, B.; Downing, J.A.; Petocz, P.; Brand-Miller, J.; Bryden, W.L. Metabolic fate of intravenously administered N-acetylneuraminic acid-6-14C in newborn piglets. Asia Pac. J. Clin. Nutr. 2007, 16, 110–115. [Google Scholar]
- Bode, L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology 2012, 22, 1147–1162. [Google Scholar] [CrossRef] [Green Version]
- Wang, B. Sialic Acid Is an Essential Nutrient for Brain Development and Cognition. Annu. Rev. Nutr. 2009, 29, 177–222. [Google Scholar] [CrossRef] [PubMed]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.S. Emotional behavior in the rat. I. Defecation and urination as measures of individual differences in emotionality. J. Comp. Psychol. 1934, 18, 385–403. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Howlett, D.R.; Bowler, K.; Soden, P.E.; Riddell, D.; Davis, J.B.; Richardson, J.C.; Burbidge, S.A.; Gonzalez, M.I.; Irving, E.A.; Lawman, A. Abeta deposition and related pathology in an APP x PS1 transgenic mouse model of Alzheimer’s disease. Histol. Histopathol. 2008, 23, 67–76. [Google Scholar]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Stieler, J.T.; Bullmann, T.; Kohl, F.; Toien, O.; Bruckner, M.K.; Hartig, W.; Barnes, B.M.; Arendt, T. The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS ONE 2011, 6, e14530. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Li, C.; Wu, H.; Feng, X.; Su, Q.; Li, S.; Zhang, L.; Yew, D.T.; Cho, E.Y.; Sha, O. Propidium iodide (PI) stains Nissl bodies and may serve as a quick marker for total neuronal cell count. Acta Histochem. 2015, 117, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.; Pinto, X.; Munoz, A.; Zuniga, M.; Rubies-Prat, J.; Pallardo, L.F.; Masana, L.; Mangas, A.; Hernandez-Mijares, A.; Gonzalez-Santos, P.; et al. Lipoprotein ratios: Physiological significance and clinical usefulness in car-diovascular prevention. Vasc. Health Risk Manag. 2009, 5, 757–765. [Google Scholar] [PubMed]
- Walldius, G.; Jungner, I. Is there a better marker of cardiovascular risk than LDL cholesterol? Apolipoproteins B and A-I–new risk factors and targets for therapy. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 565–571. [Google Scholar] [CrossRef]
- Wang, X.; Rader, D.J. Molecular regulation of macrophage reverse cholesterol transport. Curr. Opin. Cardiol. 2007, 22, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Giambattistelli, F.; Bucossi, S.; Salustri, C.; Panetta, V.; Mariani, S.; Siotto, M.; Ventriglia, M.; Vernieri, F.; Dell’Acqua, M.L.; Cassetta, E.; et al. Effects of hemochromatosis and transferrin gene mutations on iron dyshomeostasis, liver dysfunction and on the risk of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1633–1641. [Google Scholar] [CrossRef]
- Ndrepepa, G. Uric acid and cardiovascular disease. Clin. Chim. Acta 2018, 484, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ran, J.; Jiang, T. Urea; Yang, B., Sands, J.M., Eds.; Springer: Berlin, Germany, 2014; pp. 7–29. [Google Scholar]
- Al-khateeb, E.; Althaher, A.; Al-khateeb, M.; Al-Musawi, H.; Azzouqah, O.; Al-Shweiki, S.; Shafagoj, Y. Relation between uric acid and Alzheimer’s disease in elderly Jordanians. J. Alzheimers Dis. 2015, 44, 859–865. [Google Scholar] [CrossRef] [Green Version]
- Menzie, J.; Pan, C.; Prentice, H.; Wu, J.-Y. Taurine and central nervous system disorders. Amino Acids 2014, 46, 31–46. [Google Scholar] [CrossRef]
- Frej, A.D.; Otto, G.P.; Williams, R.S.B. Tipping the scales: Lessons from simple model systems on inositol imbalance in neurological disorders. Eur. J. Cell Biol. 2017, 96, 154–163. [Google Scholar] [CrossRef]
- Gundersen, R.Y.; Vaagenes, P.; Breivik, T.; Fonnum, F.; Opstad, P.K. Glycine—An important neurotransmitter and cytoprotective agent. Acta Anaesthesiol. Scand. 2005, 49, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, A.; Upadhyay, P.; Agrawal, A.; Ilango, K.; Karmakar, D.; Singh, G.P.; Dubey, G.P. Management of cognitive determinants in senile dementia of Alzheimer’s type: Therapeutic potential of a novel polyherbal drug product. J. Clin. Investig. 2014, 34, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 9, 7–10. [Google Scholar]
- Yin, T.; Yang, L.; Liu, Y.; Zhou, X.; Sun, J.; Liu, J. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood–brain barrier permeability peptide-B6 peptide for potential use in Alzheimer’s disease. Acta Biomater. 2015, 25, 172–183. [Google Scholar] [CrossRef]
- Oliveros, E.; Vazquez, E.; Barranco, A.; Ramirez, M.; Gruart, A.; Delgado-Garcia, J.M. Sialic Acid and Sialylated Oligosaccharide Supplementation during Lactation Improves Learning and Memory in Rats. Nutrients 2018, 10, 1519. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the beta-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Annaert, W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 2000, 113, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Avila, J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006, 580, 2922–2927. [Google Scholar] [CrossRef] [Green Version]
- Karr, J.W.; Szalai, V.A. Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer’s disease amyloid-beta peptide. Biochemistry 2008, 47, 5006–5016. [Google Scholar] [CrossRef]
- Prohovnik, I.; Perl, D.P.; Davis, K.L.; Libow, L.; Lesser, G.; Haroutunian, V. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology 2006, 66, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Snyder, H.M.; Carrillo, M.C.; Fazio, S.; Kim, H.; Johns, H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimer’s Dement. 2015, 11, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Reuck, J.; Maurage, C.-A.; Deramecourt, V.; Pasquier, F.; Cordonnier, C.; Leys, D.; Bordet, R. Aging and cerebrovascular lesions in pure and in mixed neurodegenerative and vascular dementia brains: A neuropathological study. Folia Neuropathol. 2018, 56, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Surguchov, A. Caveolin: A New Link between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef]
- Sherman, C.B.; Peterson, S.J.; Frishman, W.H. Apolipoprotein A-I mimetic peptides: A potential new therapy for the prevention of atherosclerosis. Cardiol. Rev. 2010, 18, 141–147. [Google Scholar] [CrossRef]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of Human Apolipoprotein A-I Preserves Cognitive Function and Attenuates Neuroinflammation and Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, E.; Bernát, G.; Csont, T.; Ferdinandy, P.; Scheich, H.; Sántha, M. Overexpression of Human Apolipoprotein B-100 Induces Severe Neurodegeneration in Transgenic Mice. J. Proteome Res. 2008, 7, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Caramelli, P.; Nitrini, R.; Maranhao, R.; Lourenco, A.C.; Damasceno, M.C.; Vinagre, C.; Caramelli, B. Increased apolipoprotein B serum concentration in Alzheimer’s disease. Acta Neurol. Scand. 1999, 100, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Takechi, R.; Galloway, S.; Pallebage-Gamarallage, M.M.; Wellington, C.L.; Johnsen, R.D.; Dhaliwal, S.S.; Mamo, J.C. Differ-ential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-beta. Br. J. Nutr. 2010, 103, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Poljak, A.; Crawford, J.; Kochan, N.A.; Wen, W.; Cameron, B.; Lux, O.; Brodaty, H.; Mather, K.; Smythe, G.A.; et al. Plasma Apolipoprotein Levels Are Associated with Cognitive Status and Decline in a Community Cohort of Older Individuals. PLoS ONE 2012, 7, e34078. [Google Scholar] [CrossRef] [Green Version]
- Al-Khateeb, E.; Al-Zayadneh, E.; Al-Dalahmah, O.; Alawadi, Z.; Khatib, F.; Naffa, R.; Shafagoj, Y. Relation between Copper, Lipid Profile, and Cognition in Elderly Jordanians. J. Alzheimer’s Dis. 2014, 41, 203–211. [Google Scholar] [CrossRef]
- Fenili, D.; Brown, M.; Rappaport, R.; McLaurin, J. Properties of scyllo-inositol as a therapeutic treatment of AD-like pathology. J. Mol. Med. 2007, 85, 603–611. [Google Scholar] [CrossRef]
- McLaurin, J.; Kierstead, M.E.; Brown, M.E.; Hawkes, C.A.; Lambermon, M.H.L.; Phinney, A.L.; Darabie, A.A.; Cousins, J.E.; French, J.E.; Lan, M.F.; et al. Cyclohex-anehexol inhibitors of A beta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 2006, 12, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Nakato, K.; Yarimizu, J.; Yamazaki, M.; Morita, M.; Takahashi, S.; Aota, M.; Saita, K.; Doihara, H.; Sato, Y.; et al. A novel glycine transporter-1 (GlyT1) inhibitor, ASP2535 (4-3-isopropyl-5-(6-phenyl-3-pyridyl)-4H-1,2,4-triazol-4-yl-2,1,3-benz oxadiazole), improves cognition in animal models of cognitive impairment in schizophrenia and Alzheimer’s disease. Eur. J. Pharmacol. 2012, 685, 59–69. [Google Scholar] [CrossRef]
- Dawson, R.; Pelleymounter, M.A.; Cullen, M.J.; Gollub, M.; Liu, S. An age-related decline in striatal taurine is correlated with a loss of dopaminergic markers. Brain Res. Bull. 1999, 48, 319–324. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, H.V.; Yoon, J.H.; Kang, B.R.; Cho, S.M.; Lee, S.; Kim, J.Y.; Kim, J.W.; Cho, Y.; Woo, J.; et al. Taurine in drinking water recovers learning and memory in the adult APP/PS1 mouse model of Alzheimer’s disease. Sci. Rep. 2014, 4, 7467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, H.; Khan, A.; Vaibhav, K.; Khan, M.M.; Ahmad, A.; Ahmad, E.; Ahmad, A.; Tabassum, R.; Islam, F.; Safhi, M.M.; et al. Taurine ameliorates neurobehavioral, neurochemical and immunohistochemical changes in sporadic dementia of Alzheimer’s type (SDAT) caused by intracerebroventricular streptozotocin in rats. Neurol. Sci. 2013, 34, 2181–2192. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Yao, C.; Liu, F.; Xiang, W.; Zuo, Y.; Feng, K.; Lu, S.; Xiang, L.; Li, M.; Li, X.; et al. Sialic Acid Ameliorates Cognitive Deficits by Reducing Amyloid Deposition, Nerve Fiber Production, and Neuronal Apoptosis in a Mice Model of Alzheimer’s Disease. NeuroSci 2022, 3, 28-40. https://doi.org/10.3390/neurosci3010002
Xiao M, Yao C, Liu F, Xiang W, Zuo Y, Feng K, Lu S, Xiang L, Li M, Li X, et al. Sialic Acid Ameliorates Cognitive Deficits by Reducing Amyloid Deposition, Nerve Fiber Production, and Neuronal Apoptosis in a Mice Model of Alzheimer’s Disease. NeuroSci. 2022; 3(1):28-40. https://doi.org/10.3390/neurosci3010002
Chicago/Turabian StyleXiao, Min, Chuangyu Yao, Fang Liu, Wei Xiang, Yao Zuo, Kejue Feng, Shuhuan Lu, Li Xiang, Muzi Li, Xiangyu Li, and et al. 2022. "Sialic Acid Ameliorates Cognitive Deficits by Reducing Amyloid Deposition, Nerve Fiber Production, and Neuronal Apoptosis in a Mice Model of Alzheimer’s Disease" NeuroSci 3, no. 1: 28-40. https://doi.org/10.3390/neurosci3010002
APA StyleXiao, M., Yao, C., Liu, F., Xiang, W., Zuo, Y., Feng, K., Lu, S., Xiang, L., Li, M., Li, X., & Du, X. (2022). Sialic Acid Ameliorates Cognitive Deficits by Reducing Amyloid Deposition, Nerve Fiber Production, and Neuronal Apoptosis in a Mice Model of Alzheimer’s Disease. NeuroSci, 3(1), 28-40. https://doi.org/10.3390/neurosci3010002