
ATTR Variant Amyloidosis in Patients with Dysphagia


{kind=link}
{kind=link}
Abstract
:1. Introduction
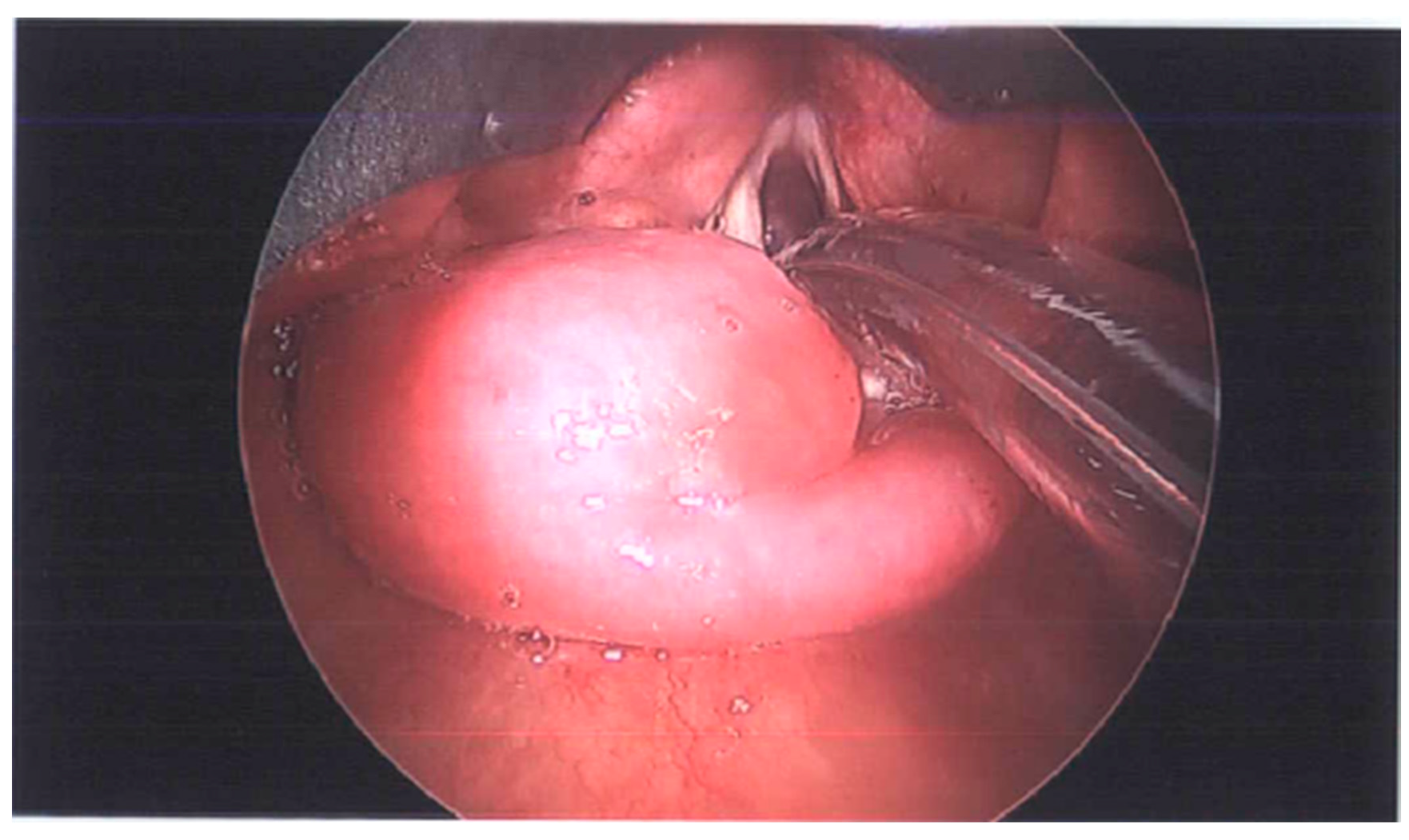
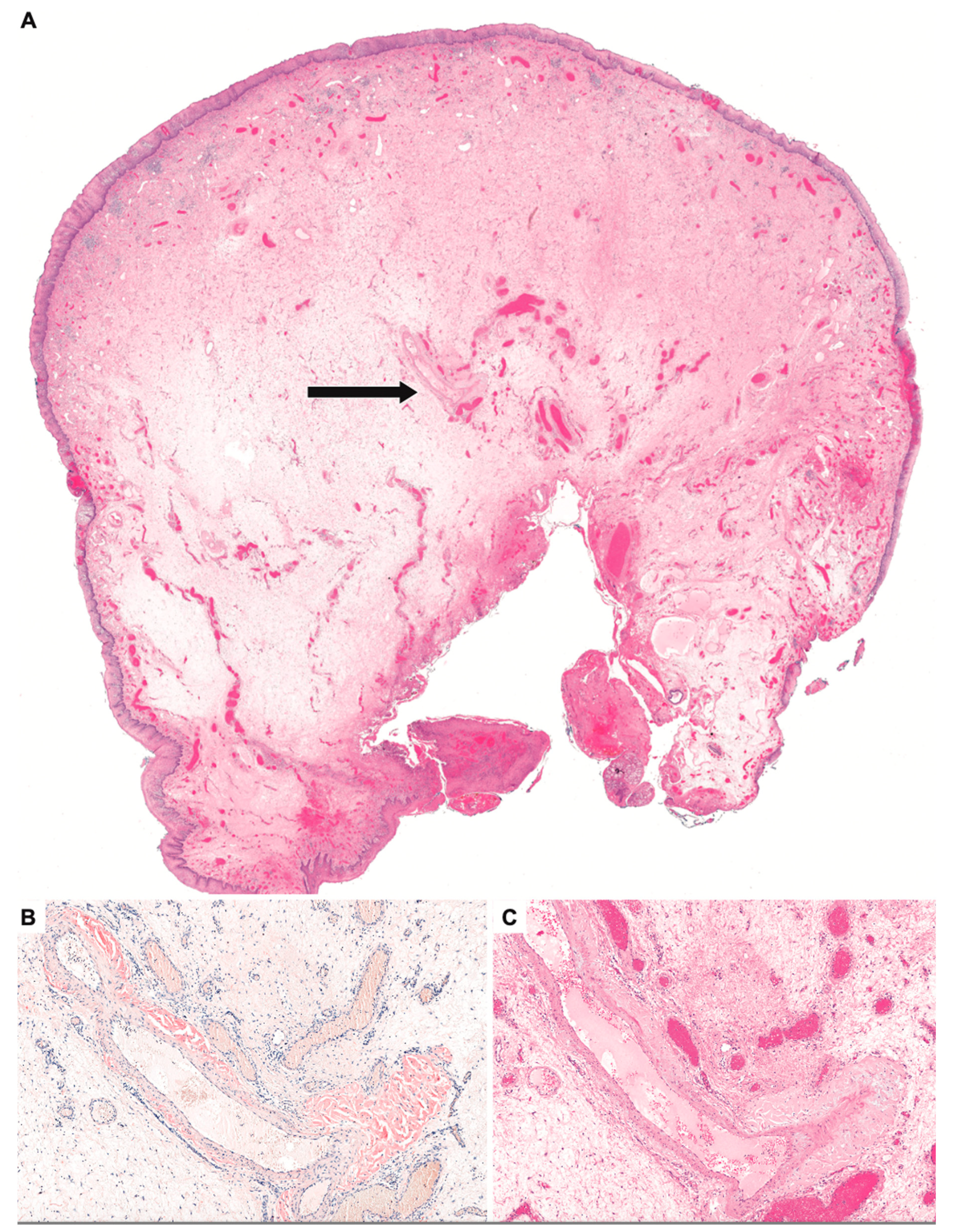
2. Detailed Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef] [PubMed]
- Rudy, S.F.; Jeffery, C.C.; Damrose, E.J. Clinical characteristics of laryngeal versus nonlaryngeal amyloidosis. Laryngoscope 2018, 128, 670–674. [Google Scholar] [CrossRef]
- Falk, R.H.; Comenzo, R.L.; Skinner, M. The Systemic Amyloidoses. N. Engl. J. Med. 1997, 337, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, B.P.C. Amyloidosis: A clinical overview. Rheum. Dis. Clin. 2013, 39, 323–345. [Google Scholar] [CrossRef] [Green Version]
- Hammami, B.; Mnejja, M.; Kallel, S.; Bouguecha, L.; Chakroun, A.; Charfeddine, I.; Ghorbel, A. Hypopharyngeal amyloidosis: A case report. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2010, 127, 83–85. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, R.; Witteles, R.; Damrose, E.; Arai, S.; Lafayette, R.A.; Schrier, S.; Afghahi, A.; Liedtke, M. More Than a Frog in the Throat: A Case Series and Review of Localized Laryngeal Amyloidosis. Arch. Otolaryngol. Neck Surg. 2012, 138, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.; Lachmann, H.; Hawkins, P.; Sandhu, G. One Hundred Cases of Localized Laryngeal Amyloidosis—Evidence for Future Management. Laryngoscope 2021, 131, E1912–E1917. [Google Scholar] [CrossRef]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Plante-Bordeneuve, V.; Schmidt, H.H.; Merlini, G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J. Am. Coll. Cardiol. 2015, 66, 2451–2466. [Google Scholar] [CrossRef] [Green Version]
- Connors, L.H.; Lim, A.; Prokaeva, T.; Roskens, V.A.; Costello, C.E. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003, 10, 160–184. [Google Scholar] [CrossRef]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef]
- Hellman, U.; Alarcon, F.; Lundgren, H.E.; Suhr, O.B.; Bonaiti-PelliÉ, C.; Planté-Bordeneuve, V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid 2008, 15, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.; Li, P.; Bae, J.Y.; Pan, S.; Dixon, R.A.F.; Liu, Q. Diagnosis and treatment of transthyretin-related amyloidosis cardiomyopathy. Clin. Cardiol. 2020, 43, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Puig-Carrion, G.D.; Reyentovich, A.; Katz, S.D. Diagnosis and treatment of heart failure in hereditary transthyretin amyloidosis. Clin. Auton. Res. 2019, 29, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Sekijima, Y.; Yazaki, M.; Tojo, K.; Yoshinaga, T.; Doden, T.; Koyama, J.; Yanagisawa, S.; Ikeda, S.-I. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016, 23, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, M.A.; Buckland, J.R.; Mason, P.; Randall, C.J.; Theaker, J. A rare case of dysphagia: Hypopharyngeal amyloidosis masquerading as a post-cricoid tumour. J. Laryngol. Otol. 2002, 116, 54–56. [Google Scholar] [CrossRef]
- Ghekiere, O.; Desuter, G.; Weynand, B.; Coche, E. Hypopharyngeal Amyloidoma. Am. J. Roentgenol. 2003, 181, 1720–1721. [Google Scholar] [CrossRef]
- Bartier, S.; Bodez, D.; Kharoubi, M.; Canouï-Poitrine, F.; Chatelin, V.; Henrion, C.; Coste, A.; Damy, T.; Béquignon, E. Pharyngo-laryngeal involvement in systemic amyloidosis with cardiac involvement: A prospective observational study. Amyloid 2019, 26, 216–224. [Google Scholar] [CrossRef]
- Ohki, M.; Kikuchi, S. Dysphagia due to systemic light chain amyloidosis revealed by videoendoscopic and videofluorographic swallowing examinations. Otolaryngol. Case Rep. 2018, 6, 4–6. [Google Scholar] [CrossRef]
- Muchtar, E.; Derudas, D.; Mauermann, M.; Liewluck, T.; Dispenzieri, A.; Kumar, S.K.; Dingli, D.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; et al. Systemic Immunoglobulin Light Chain Amyloidosis–Associated Myopathy: Presentation, Diagnostic Pitfalls, and Outcome. Mayo Clin. Proc. 2016, 91, 1354–1361. [Google Scholar] [CrossRef]
- Rubinow, A.; Burakoff, R.; Cohen, A.S.; Harris, L.D. Esophageal manometry in systemic amyloidosis. A study of 30 patients. Am. J. Med. 1983, 75, 951–956. [Google Scholar] [CrossRef]
- Abbas, Z.; Abid, S.; Rehman, N.U.; Pervez, S. Primary amyloidosis of gut presenting with dysphagia. J. Pak. Med. 1995, 45, 274–275. [Google Scholar]
- Iida, T.; Yamano, H.; Nakase, H. Systemic amyloidosis with gastrointestinal involvement: Diagnosis from endoscopic and histological views. J. Gastroenterol. Hepatol. 2018, 33, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, J. Cowan, Martha Skinner, David C. Seldin; et al. Amyloidosis of the gastrointestinal tract: A 13-year, single-center, referral experience. Haematologica 2013, 98, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Tada, S.; Lida, M.; Iwashita, A.; Matsui, T.; Fuchigami, T.; Yamamoto, T.; Yao, T.; Fujishima, M. Endoscopic and biopsy findings of the upper digestive tract in patients with amyloidosis. Gastrointest. Endosc. 1990, 36, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS—The Transthyretin Amyloidosis Outcomes Survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 2013, 29, 63–76. [Google Scholar] [CrossRef]
- Garcia, Y.; Collins, A.B.; Stone, J.R. Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Human Pathol. 2018, 72, 71–79. [Google Scholar] [CrossRef]
- Witteles, R. Cardiac Amyloidosis. 2016. Available online: https://www.acc.org/latest-in-cardiology/articles/2016/07/07/14/59/cardiac-amyloidosis (accessed on 9 May 2023).
- Conaghan, P.; Chung, D.; Vaughan, R. Recurrent laryngeal nerve palsy associated with mediastinal amyloidosis. Thorax 2000, 55, 436. [Google Scholar] [CrossRef] [Green Version]
- Batista, J.A.d.S.; Carrera, L.R.; Viriato, A.R.; Novaes, M.A.C.; de Morais, R.J.L.; Oliveira, F.T.; Marques, W.; Costa, M.C.M. Involvement of cranial nerves in ATTR Ile127Val amyloidosis. Eur. J. Med. Genet. 2022, 65, 104524. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- See, A.S.Y.; Ho, J.S.-Y.; Chan, M.Y.; Lim, Y.C.; Yeo, T.-C.; Chai, P.; Wong, R.C.; Lin, W.; Sia, C.-H. Prevalence and Risk Factors of Cardiac Amyloidosis in Heart Failure: A Systematic Review and Meta-Analysis. Hear. Lung Circ. 2022, 31, 1450–1462. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, C.H.L.; Berry, G.J.; Damrose, E.J. ATTR Variant Amyloidosis in Patients with Dysphagia. Surgeries 2023, 4, 275-282. https://doi.org/10.3390/surgeries4020028
Ng CHL, Berry GJ, Damrose EJ. ATTR Variant Amyloidosis in Patients with Dysphagia. Surgeries. 2023; 4(2):275-282. https://doi.org/10.3390/surgeries4020028
Chicago/Turabian StyleNg, Christina Hui Lee, Gerald J. Berry, and Edward J. Damrose. 2023. "ATTR Variant Amyloidosis in Patients with Dysphagia" Surgeries 4, no. 2: 275-282. https://doi.org/10.3390/surgeries4020028
APA StyleNg, C. H. L., Berry, G. J., & Damrose, E. J. (2023). ATTR Variant Amyloidosis in Patients with Dysphagia. Surgeries, 4(2), 275-282. https://doi.org/10.3390/surgeries4020028