
Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
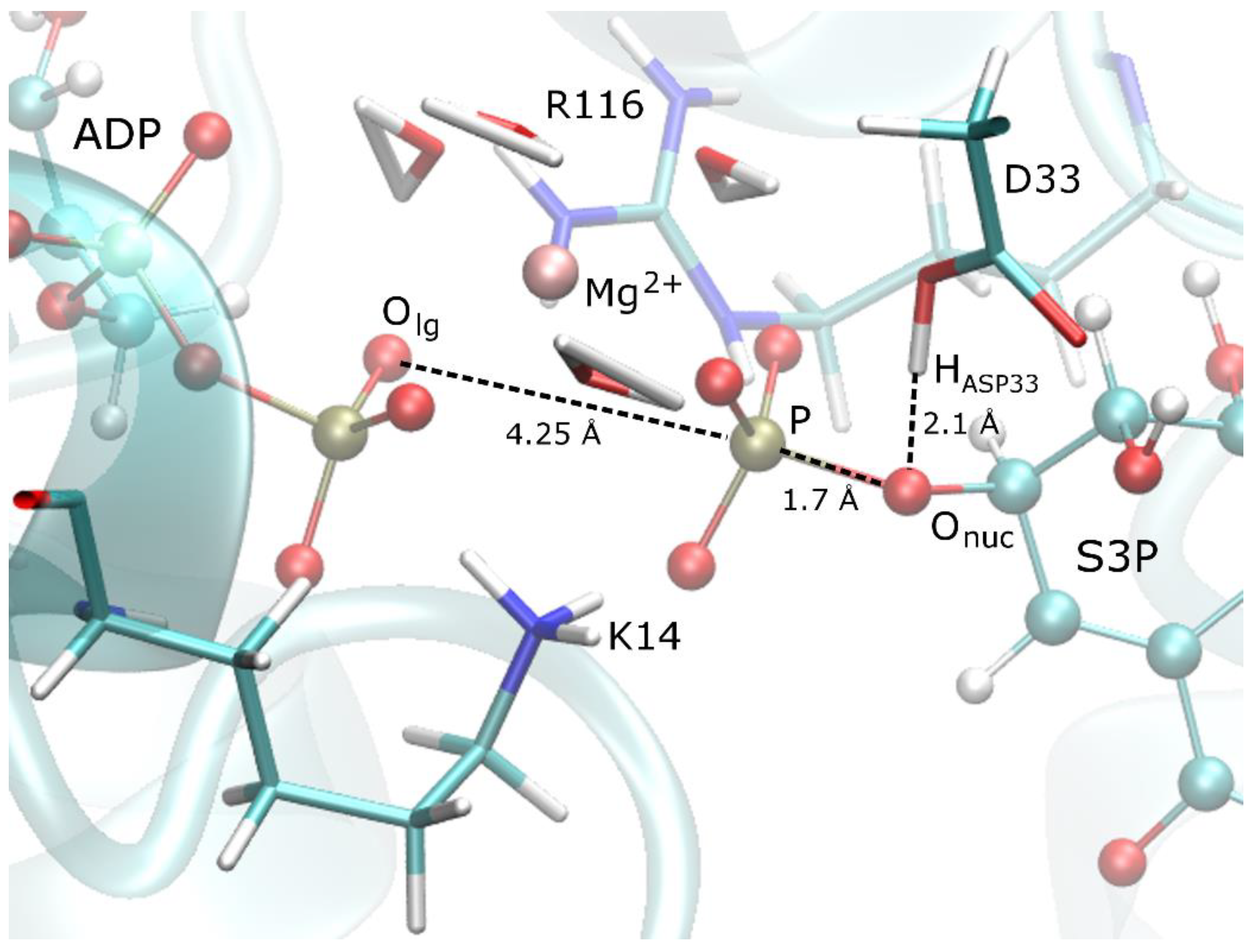
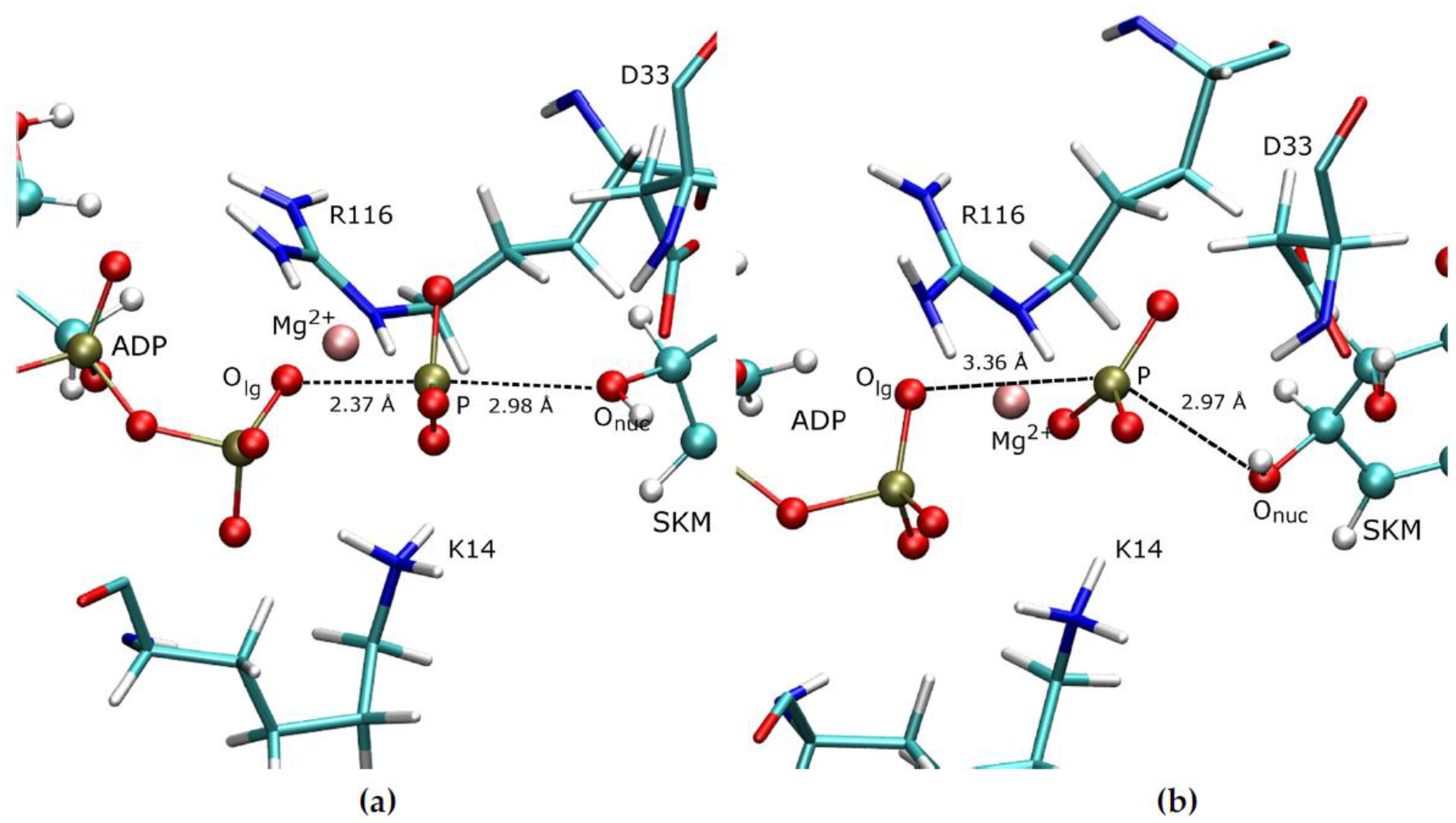
3.1. Details of the Initial Structure in the PS
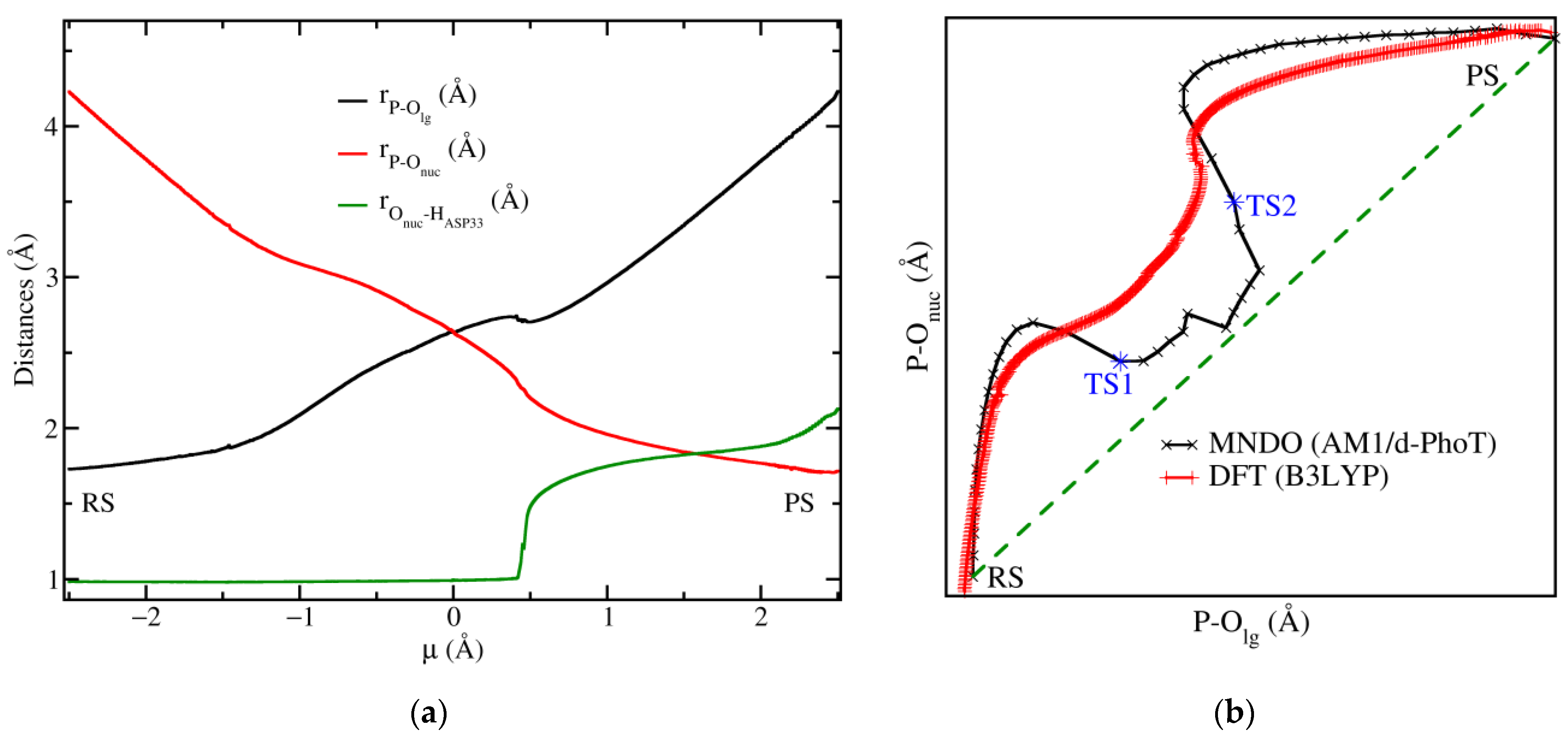
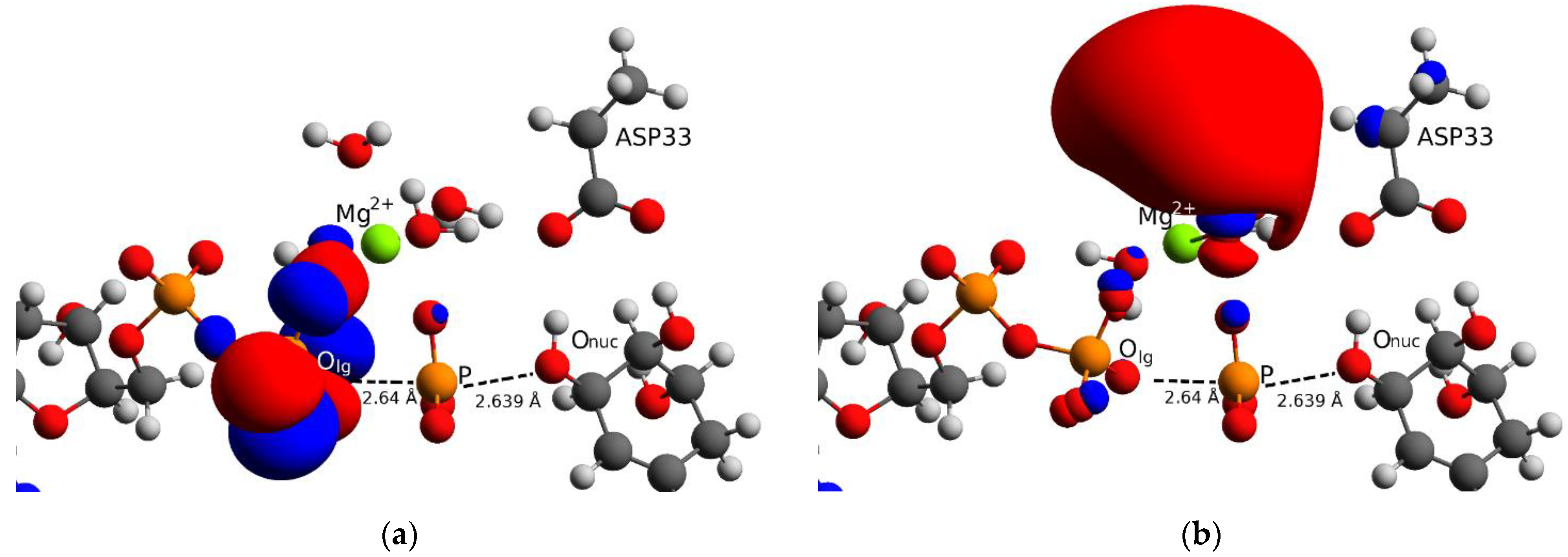
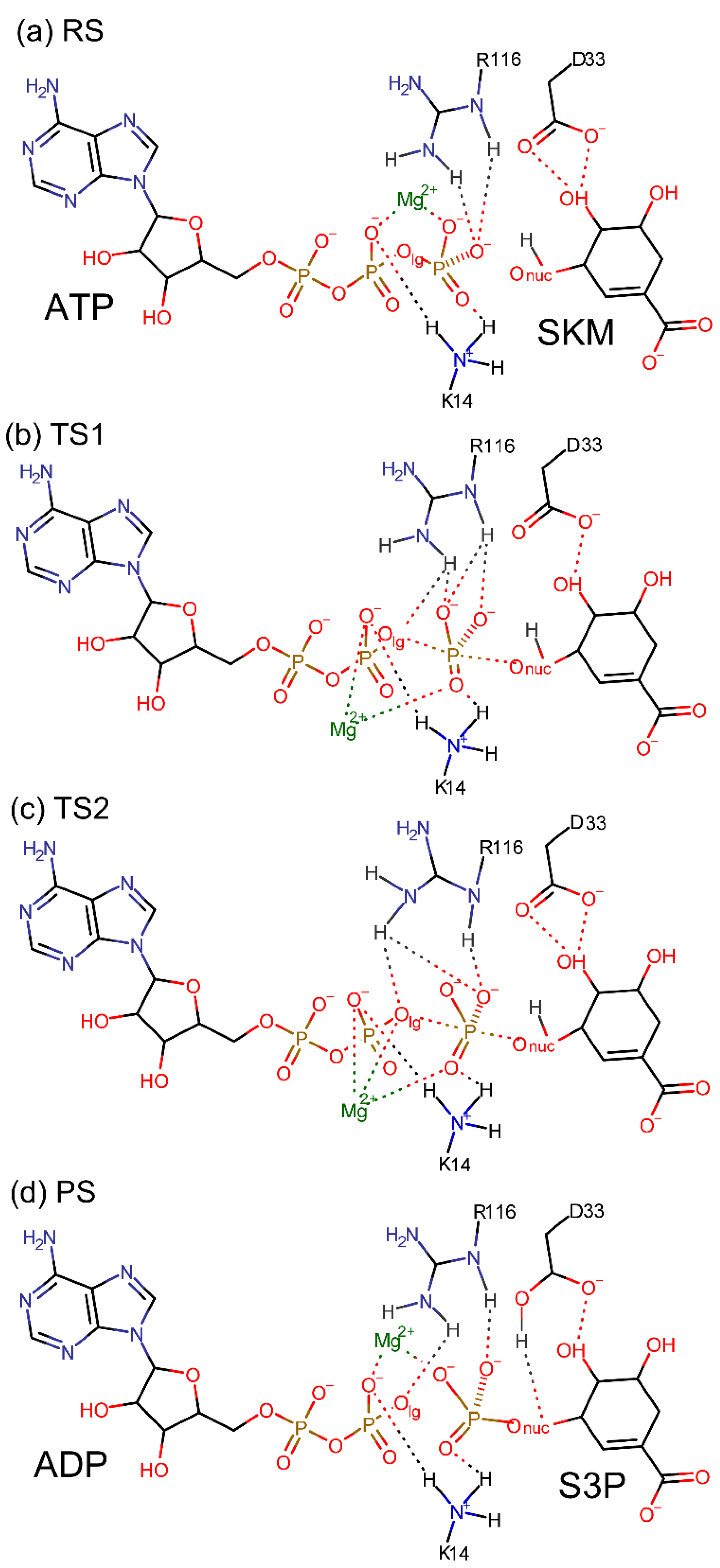
3.2. Reaction Mechanism of the Chemical Step
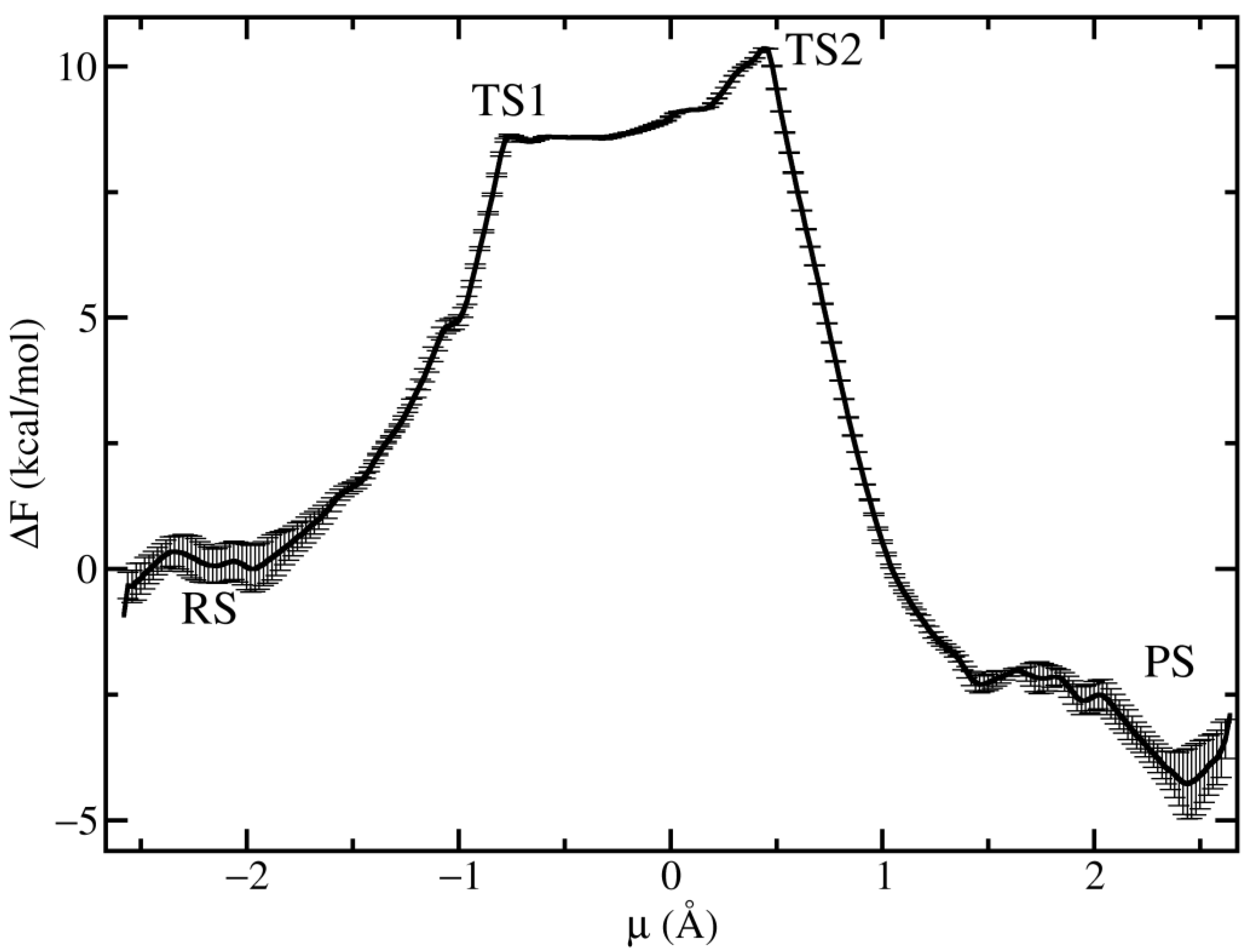
3.3. Free Energy Landscape of the Chemical Step
4. Discussion
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coracini, J.D.; de Azevedo, W.F., Jr. Shikimate kinase, a protein target for drug design. Curr. Med. Chem. 2014, 21, 592–604. [Google Scholar] [CrossRef]
- Prado, V.; Lence, E.; Vallejo, J.A.; Beceiro, A.; Thompson, P.; Hawkins, A.R.; González-Bello, C. Study of the Phosphoryl-Transfer Mechanism of Shikimate Kinase by NMR Spectroscopy. Chem. Eur. J. 2016, 22, 2758–2768. [Google Scholar] [CrossRef]
- Hartmann, M.D.; Bourenkov, G.P.; Oberschall, A.; Strizhov, N.; Bartunik, H.D. Mechanism of Phosphoryl Transfer Catalyzed by Shikimate Kinase from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 364, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-C.; Chen, Y.-F.; Wang, H.-J.; Hsu, K.-C.; Lin, S.-C.; Chen, T.-J.; Yang, J.-M.; Wang, W.-C. Structures of Helicobacter pylori Shikimate Kinase Reveal a Selective Inhibitor-Induced-Fit Mechanism. PLoS ONE 2012, 7, e33481. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.L.; Florián, J.; Warshel, A. Associative Versus Dissociative Mechanisms of Phosphate Monoester Hydrolysis: On the Interpretation of Activation Entropies. ChemPhysChem 2008, 9, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.A.; Williams, I.H.; Wilkie, J. Transition-state structural variation and mechanistic change. J. Chem. Soc. Faraday Trans. 1994, 90, 1709–1714. [Google Scholar] [CrossRef]
- Yao, J.; Wang, X.; Luo, H.; Gu, P. Understanding the Catalytic Mechanism and the Nature of the Transition State of an Attractive Drug-Target Enzyme (Shikimate Kinase) by Quantum Mechanical/Molecular Mechanical (QM/MM) Studies. Chem. Eur. J. 2018, 23, 16380–16387. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Gao, J. Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1996; pp. 119–185. [Google Scholar]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Bellinzoni, M.; Haouz, A.; Graña, M.; Munier-Lehmann, H.; Shepard, W.; Alzari, P.M. The crystal structure of Mycobacterium tuberculosis adenylate kinase in complex with two molecules of ADP and Mg2+ supports an associative mechanism for phosphoryl transfer. Protein Sci. 2006, 15, 1489–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Feig, M.; Brooks, C.L. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Banavali, N.; Foloppe, N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers 2000, 56, 257–265. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, J.A.D. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.; Gao, J.; York, D.M. An Efficient Linear-Scaling Ewald Method for Long-Range Electrostatic Interactions in Combined QM/MM Calculations. J. Chem. Theory Comput. 2005, 1, 2–13. [Google Scholar] [CrossRef]
- Nam, K. Acceleration of Ab Initio QM/MM Calculations under Periodic Boundary Conditions by Multiscale and Multiple Time Step Approaches. J. Chem. Theory Comput. 2014, 10, 4175–4183. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Rev. E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Gao, J.; Amara, P.; Alhambra, C.; Field, M.J. A Generalized Hybrid Orbital (GHO) Method for the Treatment of Boundary Atoms in Combined QM/MM Calculations. J. Phys. Chem. A 1998, 102, 4714–4721. [Google Scholar] [CrossRef]
- Nam, K.; Cui, Q.; Gao, J.; York, D.M. Specific Reaction Parametrization of the AM1/d Hamiltonian for Phosphoryl Transfer Reactions: H, O, and P Atoms. J. Chem. Theory Comput. 2007, 3, 486–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklasson, A.M.N.; Steneteg, P.; Odell, A.; Bock, N.; Challacombe, M.; Tymczak, C.J.; Holmström, E.; Zheng, G.; Weber, V. Extended Lagrangian Born–Oppenheimer molecular dynamics with dissipation. J. Chem. Phys. 2009, 130, 214109. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Niklasson, A.M.N.; Karplus, M. Lagrangian formulation with dissipation of Born-Oppenheimer molecular dynamics using the density-functional tight-binding method. J. Chem. Phys. 2011, 135, 044122. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Li, Q.; Fan, S.; Li, X.; Jin, Y.; He, W.; Zhou, J.; Cen, S.; Yang, Z. Insights into the Phosphoryl Transfer Mechanism of Human Ubiquitous Mitochondrial Creatine Kinase. Sci. Rep. 2016, 6, 38088. [Google Scholar] [CrossRef]
- Mildvan, A.S. Mechanisms of signaling and related enzymes. Proteins Struct. Funct. Bioinform. 1997, 29, 401–416. [Google Scholar] [CrossRef]
- Krell, T.; Maclean, J.; Boam, D.J.; Cooper, A.; Resmini, M.; Brocklehurst, K.; Kelly, S.M.; Price, N.C.; Lapthorn, A.J.; Coggins, J.R. Biochemical and X-ray crystallographic studies on shikimate kinase: The important structural role of the P-loop lysine. Protein Sci. 2001, 10, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Jencks, W.P. A primer for the Bema Hapothle. An empirical approach to the characterization of changing transition-state structures. Chem. Rev. 1985, 85, 511–527. [Google Scholar] [CrossRef]
- More, O.; Ferrall, R.A. Relationships between E2 and E1cB mechanisms of β-elimination. J. Chem. Soc. B 1970, 274, 77. [Google Scholar] [CrossRef]
- Rosenstock, H.M.; Wallenstein, M.B.; Wahrhaftig, A.L.; Eyring, H. Absolute Rate Theory for Isolated Systems and the Mass Spectra of Polyatomic Molecules. Proc. Natl. Acad. Sci. USA 1952, 38, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojeda-May, P. Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica 2021, 1, 334-343. https://doi.org/10.3390/biophysica1030025
Ojeda-May P. Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica. 2021; 1(3):334-343. https://doi.org/10.3390/biophysica1030025
Chicago/Turabian StyleOjeda-May, Pedro. 2021. "Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods" Biophysica 1, no. 3: 334-343. https://doi.org/10.3390/biophysica1030025
APA StyleOjeda-May, P. (2021). Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica, 1(3), 334-343. https://doi.org/10.3390/biophysica1030025