
Insights into the Substrate Uptake Mechanism of Mycobacterium Tuberculosis Ribose 5-Phosphate Isomerase and Perspectives on Drug Development

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Nuclear Magnetic Resonance
2.3. Site-Directed Docking
2.4. Molecular Dynamics Simulation
3. Results
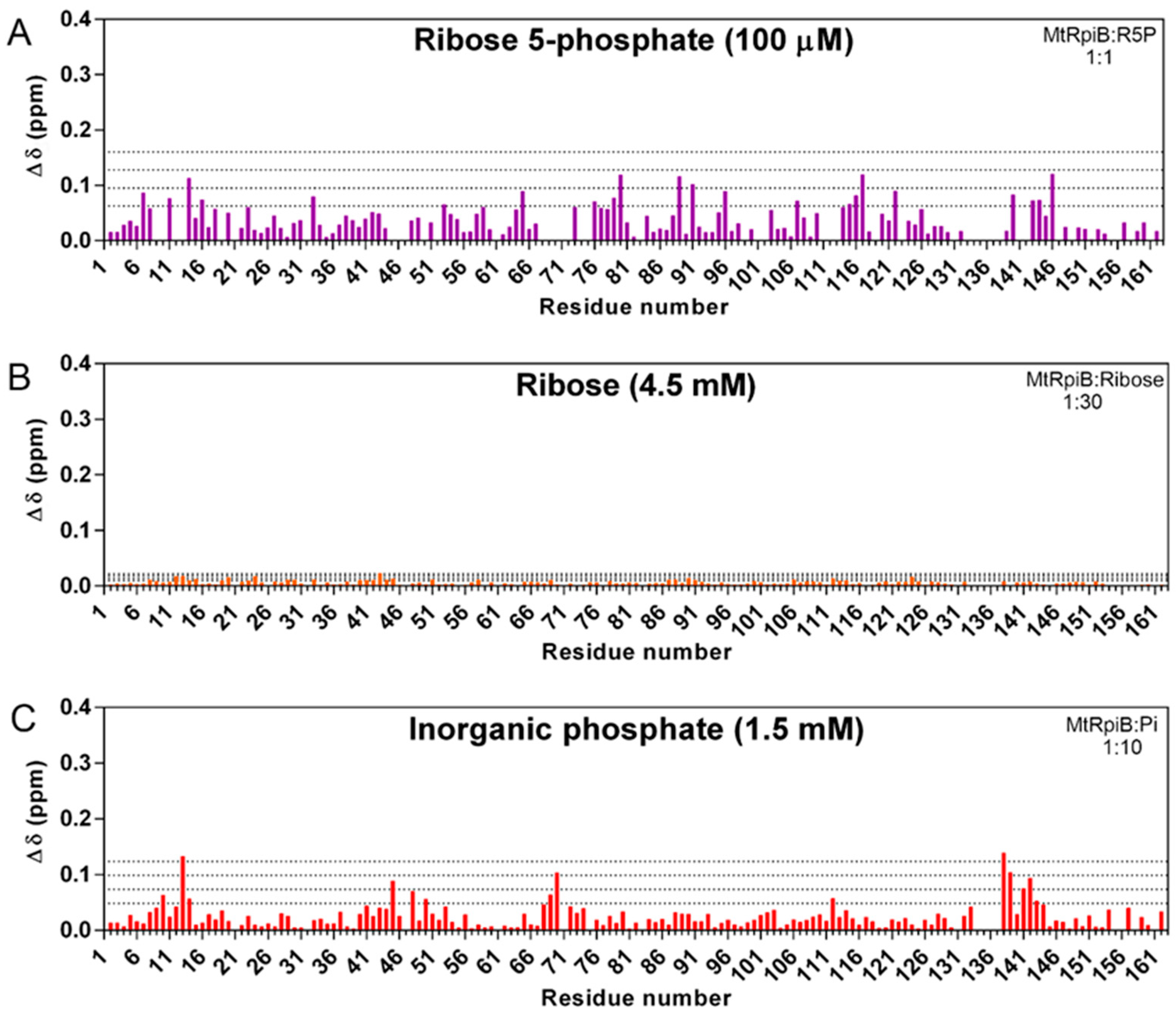
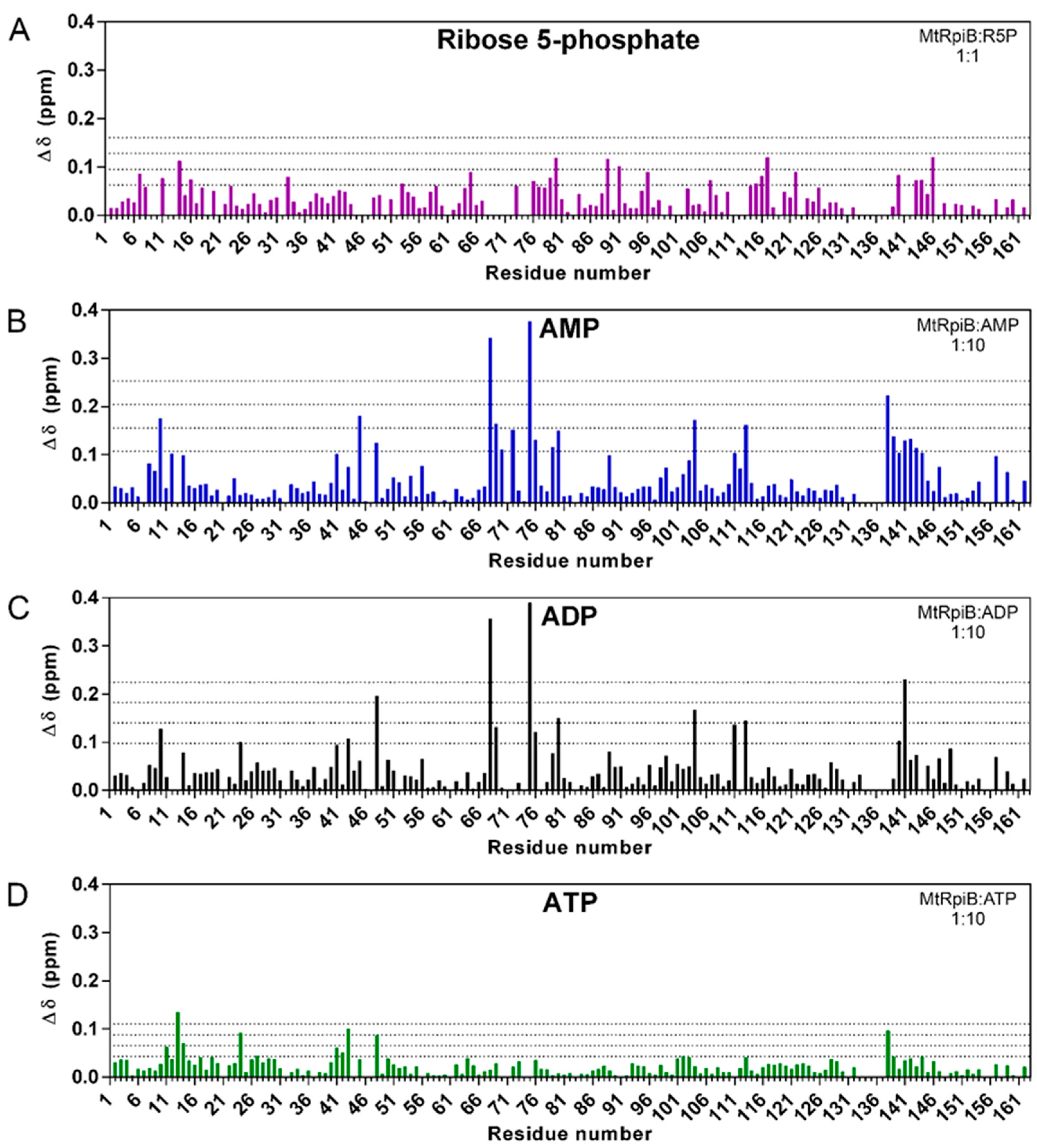
3.1. The Phosphate Group Is Essential to the Interaction
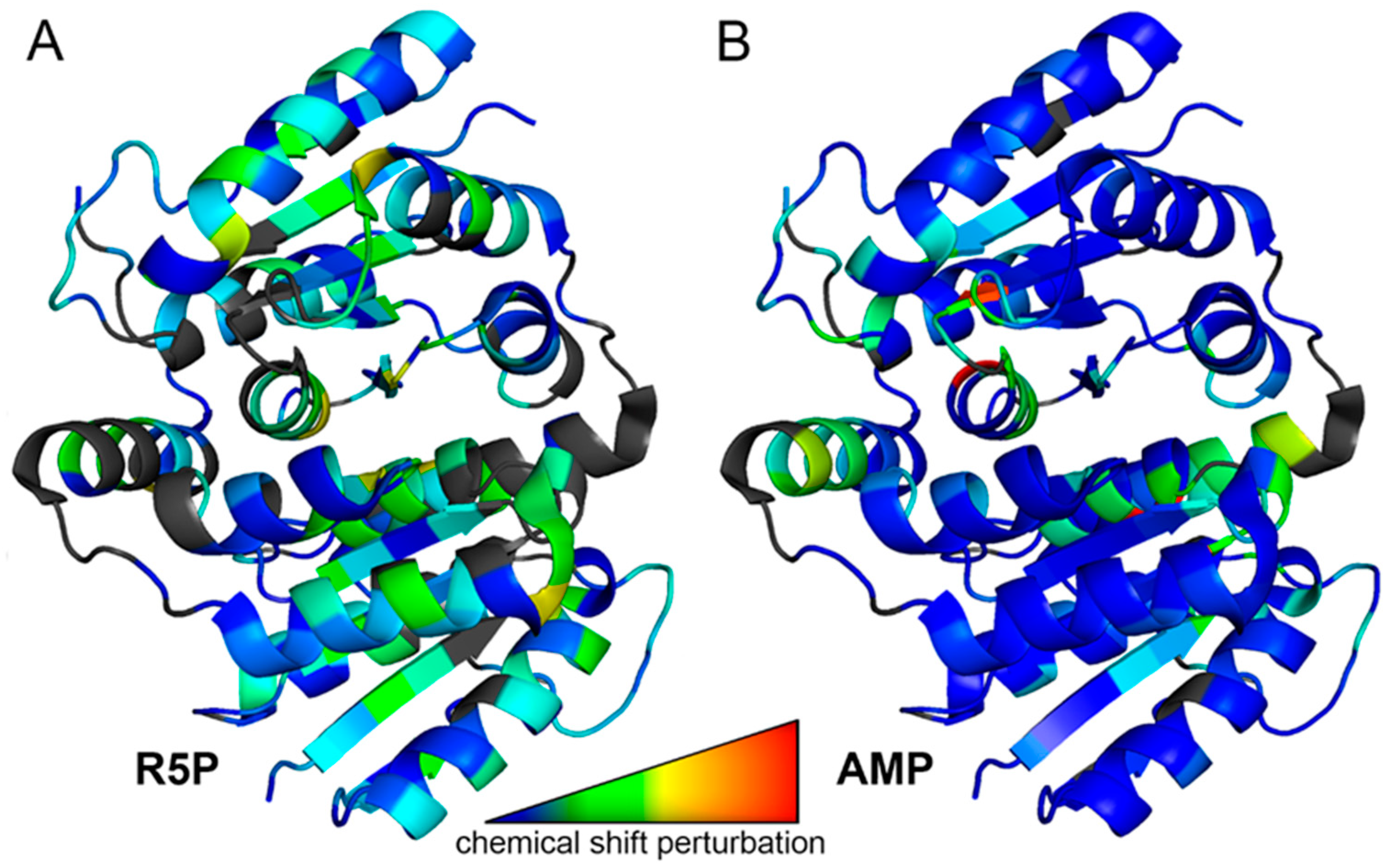
3.2. MtRpiB Interacts with AMP Derivatives
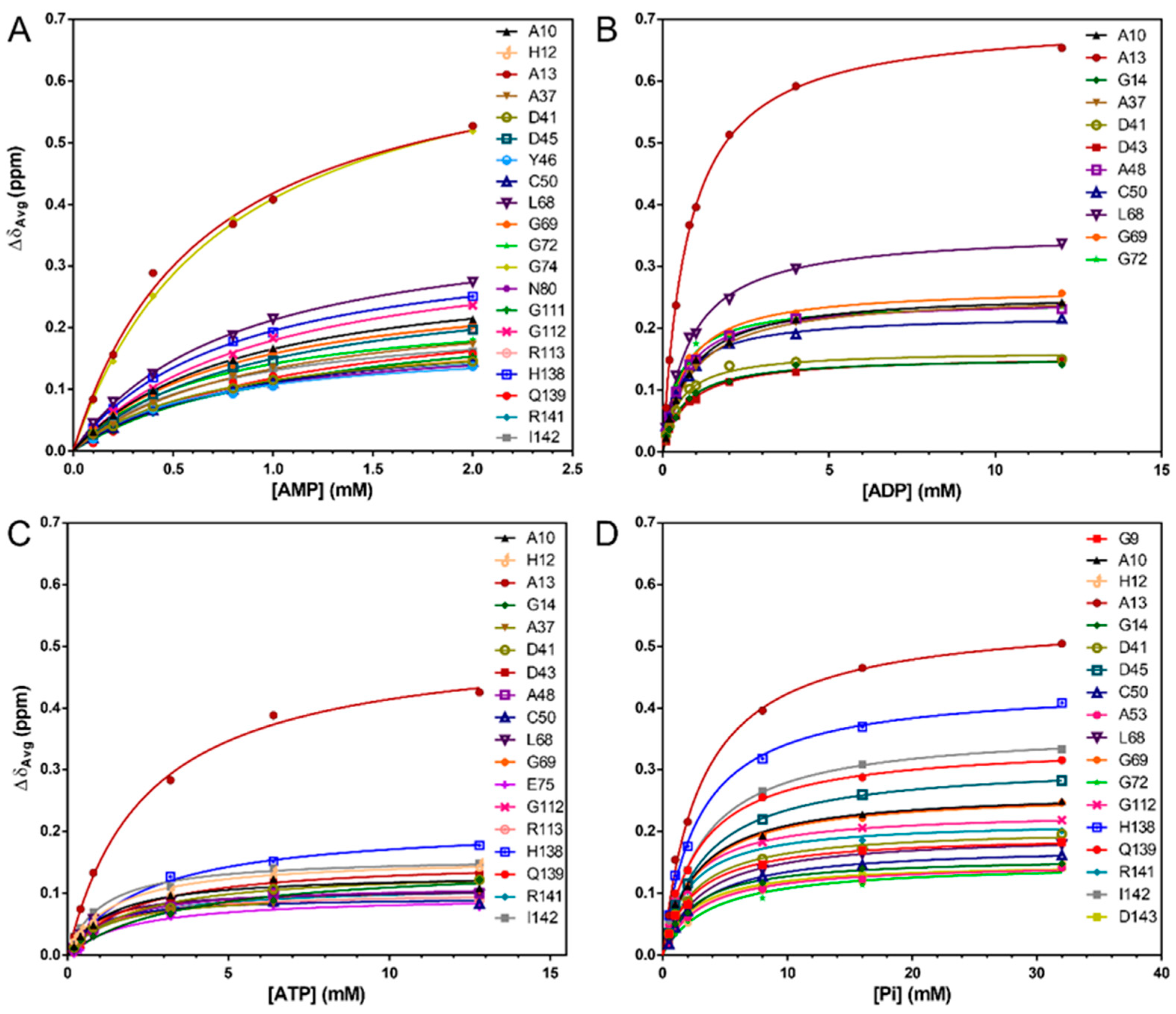
3.3. The Interaction of Nucleotides with MtRpiB Has a Low Affinity
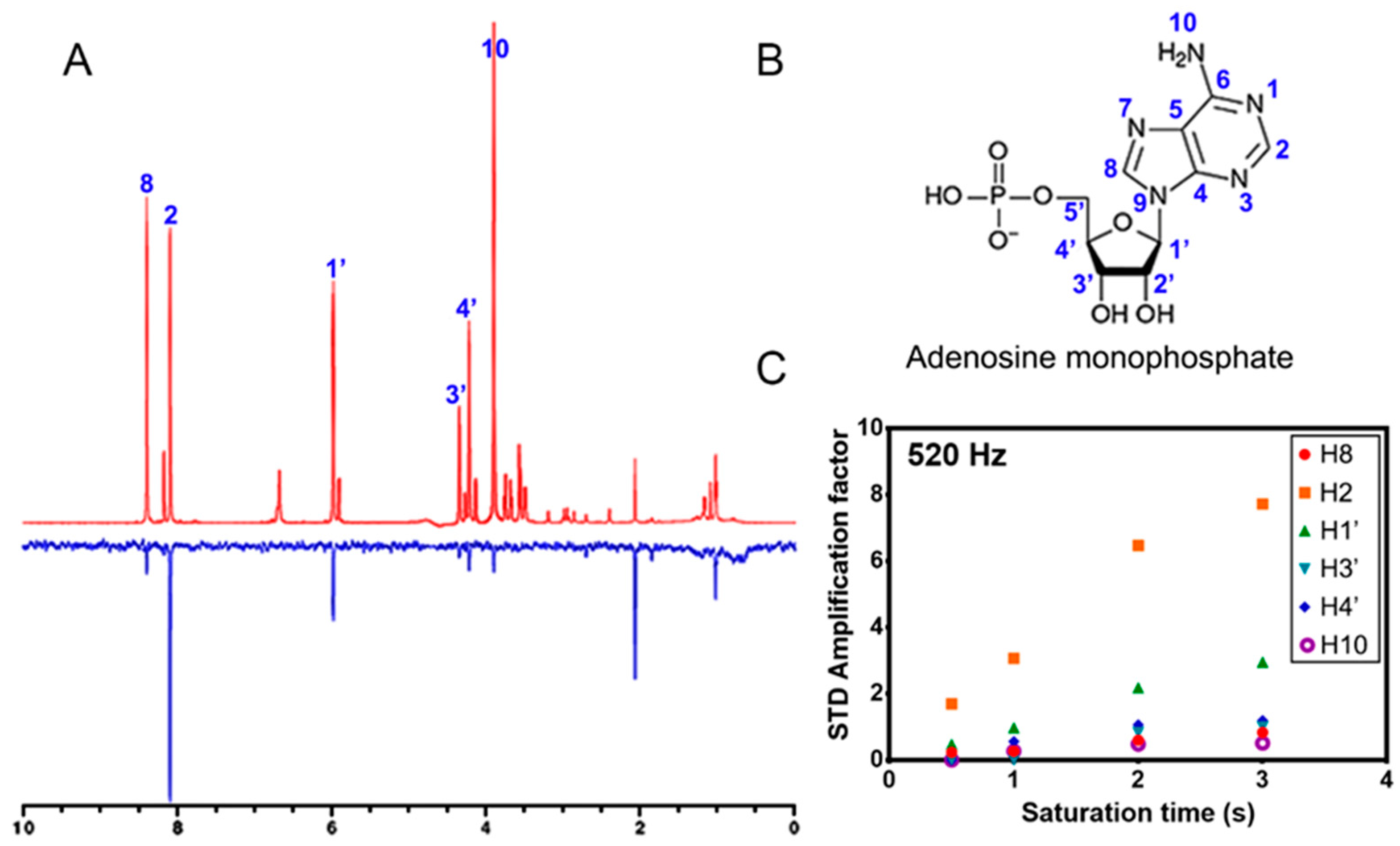
3.4. AMP Interaction with MtRpiB by Saturation Transfer Difference (STD)
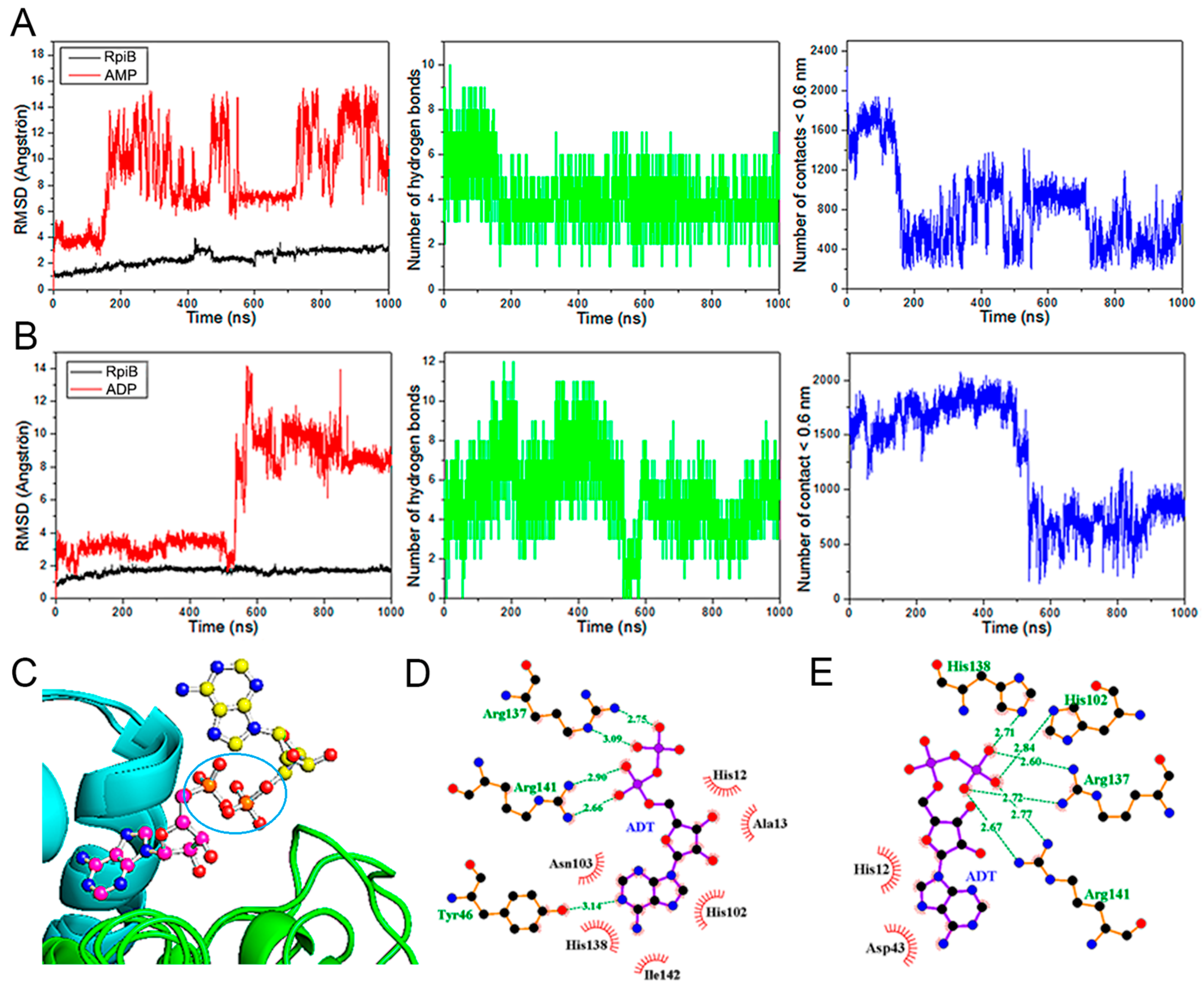
3.5. Different Forces Drive the MtRpiB Interaction with R5P and AMP
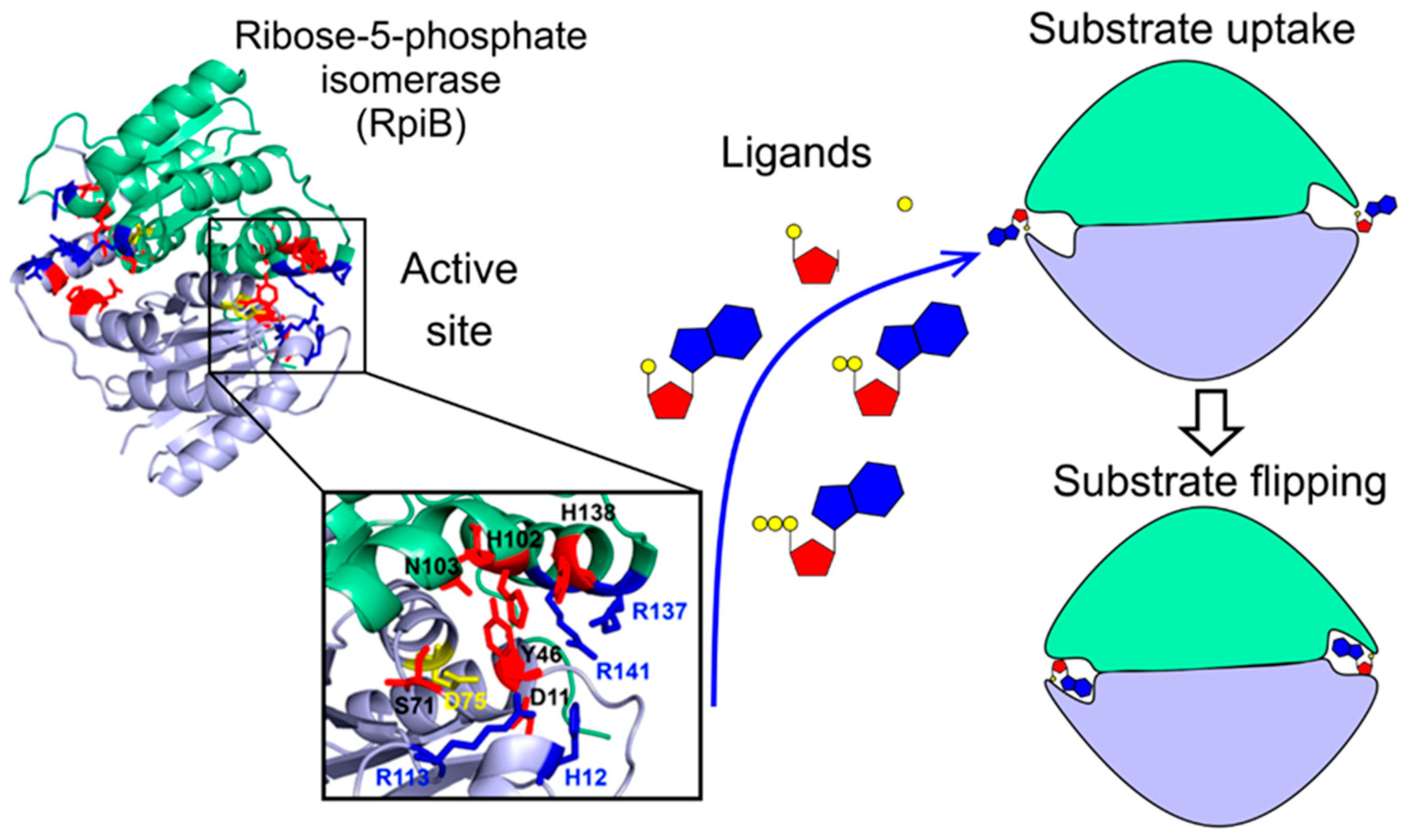
3.6. A Flipping Mechanism Is Necessary for Ligand Internalization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandner, A.; Ngo, K.; Sager, C.; Scheer, F.; Daude, M.; Diederich, W.; Heine, A.; Klebe, G. Which properties allow ligands to open and bind to the transient binding pocket of human aldose reductase? Biomolecules 2021, 11, 1387. [Google Scholar] [CrossRef] [PubMed]
- Sultana, K.N.; Srivastava, S.K. Structural and molecular dynamics of ammonia transport in Staphylococcus aureus NH 3 -dependent NAD synthetase. Int. J. Biol. Macromol. 2022, 203, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Villali, J.; Kern, D. Choreographing an enzyme’s dance. Curr. Opin. Chem. Biol. 2010, 14, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-G.; guAngdersson, C.; Savchenko, A.; Skarina, T.; Etvdokimova, E.; Beasley, S.; Arrowsmith, C.; Edwards, A.M.; Joachimiak, A.; Mowbray, S.L. Structure of Escherichia coli ribose-5-phosphate isomerase: A ubiquitous enzyme of the pentose phosphate pathway and the Calvin cycle. Structure 2003, 11, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, K.I.; Sørensen, K.; Hove-Jensen, B. Ribose catabolism of Escherichia coli: Characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J. Bacteriol. 1996, 178, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Faria, J.; Loureiro, I.; Santarém, N.; Cecílio, P.; Macedo-Ribeiro, S.; Tavares, J.; Cordeiro-Da-Silva, A. Disclosing the essentiality of ribose-5-phosphate isomerase B in Trypanosomatids. Sci. Rep. 2016, 6, 26937. [Google Scholar] [CrossRef] [Green Version]
- DeJesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Capriles, P.V.; Baptista, L.P.R.; Guedes, I.A.; Guimarães, A.C.R.; Custódio, F.L.; Alves-Ferreira, M.; Dardenne, L.E. Structural modeling and docking studies of ribose 5-phosphate isomerase from Leishmania major and Homo sapiens: A comparative analysis for Leishmaniasis treatment. J. Mol. Graph. Model. 2015, 55, 134–147. [Google Scholar] [CrossRef]
- Roos, A.K.; Andersson, C.; Bergfors, T.; Jacobsson, M.; Karlén, A.; Unge, T.; Jones, T.; Mowbray, S.L. Competitive inhibitors of Mycobacterium tuberculosis ribose-5-phosphate isomerase B reveal new information about the reaction mechanism. J. Biol. Chem. 2005, 280, 6416–6422. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.K.; Andersson, C.; Bergfors, T.; Jacobsson, M.; Karlén, A.; Unge, T.; Jones, T.; Mowbray, S.L. Mycobacterium tuberculosis ribose-5-phosphate isomerase has a known fold, but a novel active site. J. Mol. Biol. 2004, 335, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, I.; Faria, J.; Clayton, C.; Macedo-Ribeiro, S.; Santarém, N.; Roy, N.; Cordeiro-Da-Siva, A.; Tavares, J. Ribose 5-phosphate isomerase B knockdown compromises Trypanosoma brucei bloodstream form infectivity. PLoS Negl. Trop. Dis. 2015, 9, e3430. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.K.; Mariano, S.; Kowalinski, E.; Salmon, L.; Mowbray, S.L. Isomerase B from Escherichia coli is Also a Functional D -Allose-6-Phosphate Isomerase, While the Mycobacterium tuberculosis Enzyme is Not D-Ribose-5-Phosphate. J. Mol. Biol. 2008, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariano, S.; Roos, A.; Mowbray, S.; Salmon, L. Competitive inhibitors of type B ribose 5-phosphate isomerases: Design, synthesis and kinetic evaluation of new D -allose and D -allulose 6-phosphate derivatives. Carbohydr. Res. 2009, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Bartkevihi, L.; Martins, B.; de Oliveira, D.M.P.; Pires, J.R.M.; Anobom, C.D.; Almeida, F.C.L. Backbone assignment of ribose-5-phosphate isomerase of Mycobacterium tuberculosis (MtRpiB). Biomol. NMR Assign. 2020, 14, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; James, T. Detecting Ligand Binding to a Small RNA Target via Saturation Transfer Difference NMR Experiments in D2O and H2O. J. Am. Chem. Soc. 2002, 124, 13376–13377. [Google Scholar] [CrossRef] [PubMed]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-transfer difference (STD) NMR: A simple and fast method for ligand screening and characterization of protein binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Linge, J.P.; Williams, M.A.; Spronk, C.A.E.M.; Bonvin, A.M.J.J.; Nilges, M. Refinement of protein structures in explicit solvent. Proteins Struct. Funct. Bioinforma. 2003, 50, 496–506. [Google Scholar] [CrossRef] [Green Version]
- Delano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ 1 and χ 2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Benoni, R.; Pertinhez, T.; Spyrakis, F.; Davalli, S.; Pellegrino, S.; Paredi, G.; Pezzotti, A.; Bettati, S.; Campanini, B.; Mozzarelli, A. Structural insight into the interaction of O- acetylserine sulfhydrylase with competitive, peptidic inhibitors by saturation transfer difference-NMR. FEBS Lett. 2016, 590, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Sá, J.M.; Piloto, J.V.; Cilli, E.M.; Tasic, L.; Fossey, M.A.; Almeida, F.C.L.; Souza, F.P.; Caruso, P. Hesperetin targets the hydrophobic pocket of the nucleoprotein/phosphoprotein binding site of human respiratory syncytial virus. J. Biomol. Struct. Dyn. 2020, 40, 2156–2168. [Google Scholar] [CrossRef]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of amp-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.N.; Dunaway-Mariano, D. Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 2009, 19, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, G.R.J.; Campbell, A.S. Phosphonates as mimics of phosphate biomolecules: ab initio calculations on tetrahedral ground states and pentacoordinate intermediates for phosphoryl transfer. J. Org. Chem. 1993, 58, 2272–2281. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartkevihi, L.; Caruso, Í.P.; Martins, B.; Pires, J.R.M.; Oliveira, D.M.P.; Anobom, C.D.; Almeida, F.C.L. Insights into the Substrate Uptake Mechanism of Mycobacterium Tuberculosis Ribose 5-Phosphate Isomerase and Perspectives on Drug Development. Biophysica 2023, 3, 139-157. https://doi.org/10.3390/biophysica3010010
Bartkevihi L, Caruso ÍP, Martins B, Pires JRM, Oliveira DMP, Anobom CD, Almeida FCL. Insights into the Substrate Uptake Mechanism of Mycobacterium Tuberculosis Ribose 5-Phosphate Isomerase and Perspectives on Drug Development. Biophysica. 2023; 3(1):139-157. https://doi.org/10.3390/biophysica3010010
Chicago/Turabian StyleBartkevihi, Leonardo, Ícaro P. Caruso, Bruna Martins, José R. M. Pires, Danielle M. P. Oliveira, Cristiane Dinis Anobom, and Fabio C. L. Almeida. 2023. "Insights into the Substrate Uptake Mechanism of Mycobacterium Tuberculosis Ribose 5-Phosphate Isomerase and Perspectives on Drug Development" Biophysica 3, no. 1: 139-157. https://doi.org/10.3390/biophysica3010010
APA StyleBartkevihi, L., Caruso, Í. P., Martins, B., Pires, J. R. M., Oliveira, D. M. P., Anobom, C. D., & Almeida, F. C. L. (2023). Insights into the Substrate Uptake Mechanism of Mycobacterium Tuberculosis Ribose 5-Phosphate Isomerase and Perspectives on Drug Development. Biophysica, 3(1), 139-157. https://doi.org/10.3390/biophysica3010010