3.1. Refinement of the TsaBDE Complex from T. maritima and Qri7 from Yeast
To obtain reliable results for our docking studies, aimed at elucidating the tRNA binding mode, we needed to utilize the best models for the structures of the TsaD-like elements. Only complete and well-refined structures can provide the basis for drawing conclusions based on docking. Therefore, we initiated the review of quality and the refinement of selected structures.
The coordinates and the associated diffraction data of
T. maritima TsaBDE proteins in the 2:2:2 complex and Qri7 from yeast were retrieved from PDB (6FPE, 3WUH) [
2,
16]. The refinements have been completed in several macrocycles, consisting of a round of visual inspection and manual modeling/correcting of the structures, followed by ~20 cycles of LSQ refinement in Refmac 5.8. The structures were improved by modeling the difference in electron densities, by placing or removing protein fragments, and by repositioning other fragments. This procedure led to improvements in the overall completeness of the model. The main descriptors like R and Rfree improved, while the resulting model maintained good stereochemistry despite modeling fragments with weaker electron densities at lower contouring levels.
The refined structure showed improvements to the initial model in the backbone placement (
Figure S1) as well as many side-chains as measured by comparison to other models, particularly [
11]. The final model of the refined TsaBDE complex (6NAK) contains more than 50 newly placed residues and more than 190 residues in new positions. This constitutes more than 15% of the full sequence of the original structure. The newly refined elements improved the standard R factor (0.19 from the original 0.22) while leaving the Rfree at the same level. The detailed findings are described in the
Supplement.
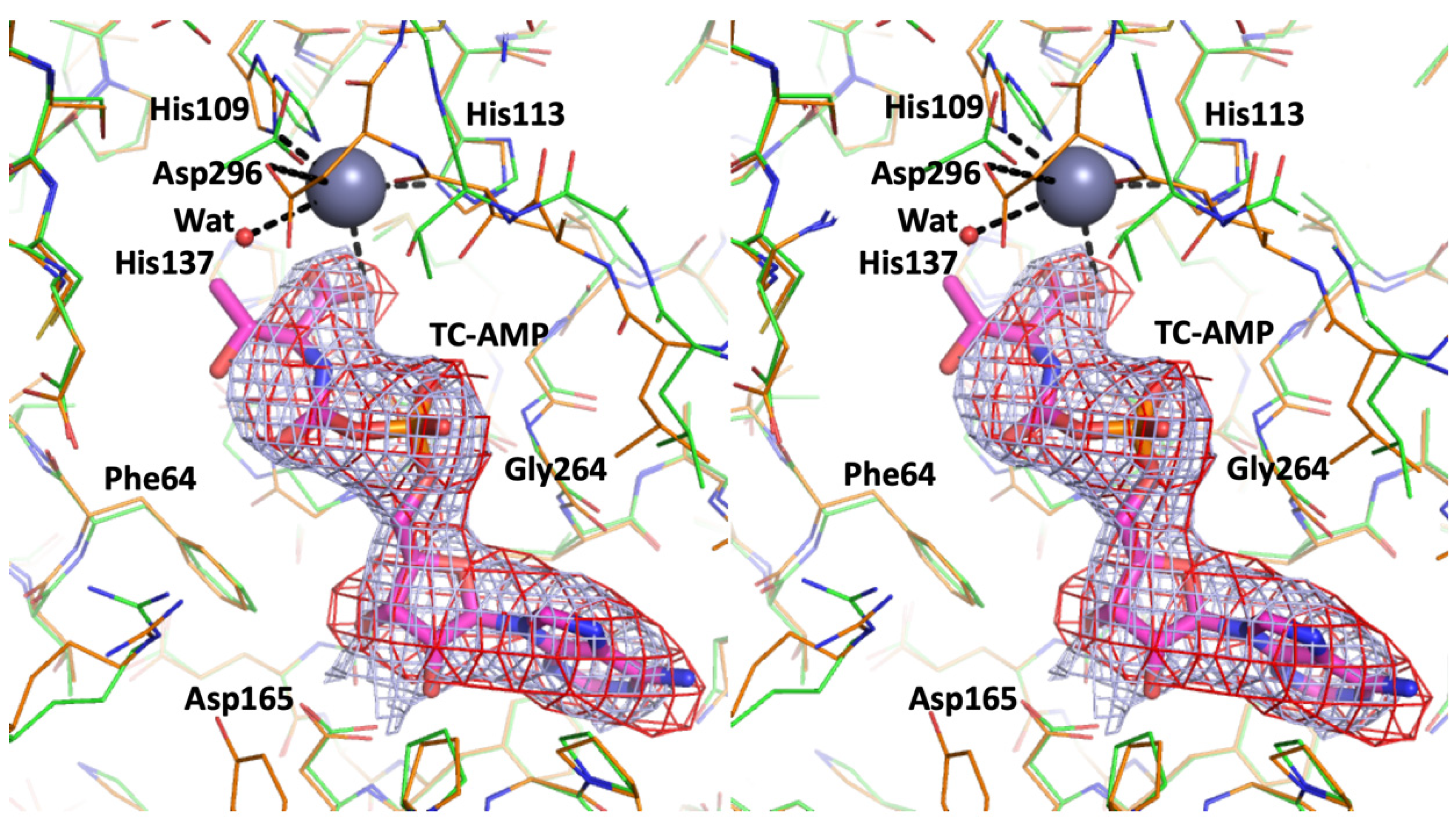
All the improvements lead to two new elements that deserve mentioning as they contribute to a better understanding of the function of this complex. The electron densities suggested the presence of a substrate, TC-AMP (
Figure 1), in both active sites of TsaD subunits instead of the originally placed PEG and glycerol. Additionally, binding of the TsaE component caused a significant reorganization of the 40–50 loop in both TsaD subunits (
Figure 2) that was not modeled in the original determination. This conformational transition in TsaD, caused by the binding of TsaE, must be associated with the alternative/antagonistic binding of tRNA or TsaC to the active site, as described in [
11]. The TsaE element therefore plays a controlling and releasing function in opposition to the suggested role of causing disorder at the active site [
2]. The refined TC-AMP at the active site sheds light on the catalysis by the TsaD component of the t6A pathway and the transfer paths of unstable product/substrate TC-AMP from the TsaC/C2 to TsaD.
Including the substrate TC-AMP in the model improved the electron density at the active site and suggested the presence of partially occupied metal ions. Zinc ions were refined with 40% occupancy at both active sites, which was fully compatible with partially disordered binding residues, which were identified before in [
11] (His109, His113, His137, Asp296). Its coordination sphere is looser than that in 6N9A, with longer bonds to the ligands and His137 not interacting with the Zn ion. Binding of the substrate apparently caused disordering of the region 291–294 as indicated by weaker electron density. These changes resulted in a transition to a more twisted/open conformation of the TsaD that is visualized in
Figure 2. The substrate binding domain shows a noticeable displacement and rotation of 17° as compared to the reference structure of the
E. coli TsaB-TsaD heterodimer (4YDU) [
24]. The observed opening of the active site cavity (
Figure 2B) must have functional significance, as binding of the tRNA to the active site cleft would require the breathing motion to bring the A37 close to the reactive center located at the TC-AMP molecule.
The comparison of the
E. coli TsaBD complex with the
T. maritima TsaBDE complex clearly suggests that the binding of TsaE creates a conformational change that would prevent the TsaC binding or, alternatively, its expulsion from the dimeric TsaCD complex, while at the same time it opens up the TsaD active site cleft, thus promoting the tRNA binding upon dissociation from the complex. The new model of the TsaBDE complex achieves consensus with the newer model described in [
11] as well as with the corrections introduced to the recently published model [
2]. An important consequence of the new model was an altered electrostatic signature that allowed for successful docking of the tRNA. The tentative localization by X-ray crystallography of the TC-AMP also contributed to the success of this procedure by providing a key constraint for the tRNA docking. Our success in identifying the TC-AMP at the active site is clearly confirmed by the capture of the stable substrate analog in the TsaBD complex from
E. coli [
23].
The Qri7 structure was refined well, with small changes to the original model that were contained to the mobile loops 70–80 that are analogous to the loops 40–50 in TsaD. The modeling of the missing elements proved to be important in understanding the tRNA binding. The final model showed again a modest improvement in both R and Rfree for a complete model. An important change was proposed: the repacking of both subunits so they were organized in the dimer with biological relevance. Only this new dimeric form was used in subsequent docking experiments. The packing of the interface in the D-D dimer of Qri7 (6NBJ) formed a four-helical bundle reminiscent of the B-D interface in the
T. maritima complex (6NAK, 6N9A) [
11].
The models representing different complexes of the yeast KEOPS system were visually inspected, but as a result, they were only marginally changed and are not described in detail. However, appropriately superposed subunits were used to understand the conformational transitions facilitating the binding of multiple elements, such as auxiliary proteins and tRNA, and the transfer of substrates between subunits interacting during the cycle. These comparisons were also used for proposing and understanding the catalytic cycles for TsaC/C2 and TsaD and understanding the roles of individual domains in Sua5 as well as the mobility of domains in TsaD. Finally, they were indispensable in proposing the catalytic mechanisms for converting TsaC to TsaD and the transfer pathway for catalytic intermediates between TsaC and TsaD active sites, as described below.
3.2. Main Structural Changes to the DBBD Dimer Caused by Binding of the TsaE Element as Compared to Structural Changes in Qri7 and KEOPS Complex
The existing models for all components (TsaC/B/D/E) were mutually compared to the previously solved and deposited models, such as, for instance, 3ZEU [
25] for the TsaBD complex, 2A6A [
33] for the YeaZ dimer of TsaB [
34], or 1HTW for TsaE [
35]. During Qri7 modeling, both deposited models were used for comparisons (3WUH, 4K25) [
16,
36].
Recent findings shed light on the role of TsaE in controlling the activity of BD or DBBD complexes [
11]. Binding of TsaE competes with tRNA, but at the same time it primes the TsaBD complex for binding to tRNA [
4]. Luthra et al. [
4,
11] demonstrated that this general control is exerted by the ATPase activity of the E component. In Qri7 [
36], the TsaD-like element forms a TsaD homodimer by forming the four-helical bundle at the interface. Therefore, the absence of other protein components needed in other complexes for activating the dimer means that only the internal mobility of the protein controls tRNA binding. The Qri7 dimer appears to be the simplest and most basic complex to study the tRNA-TsaD interaction, therefore it should have the most distinct signature/motif for binding.
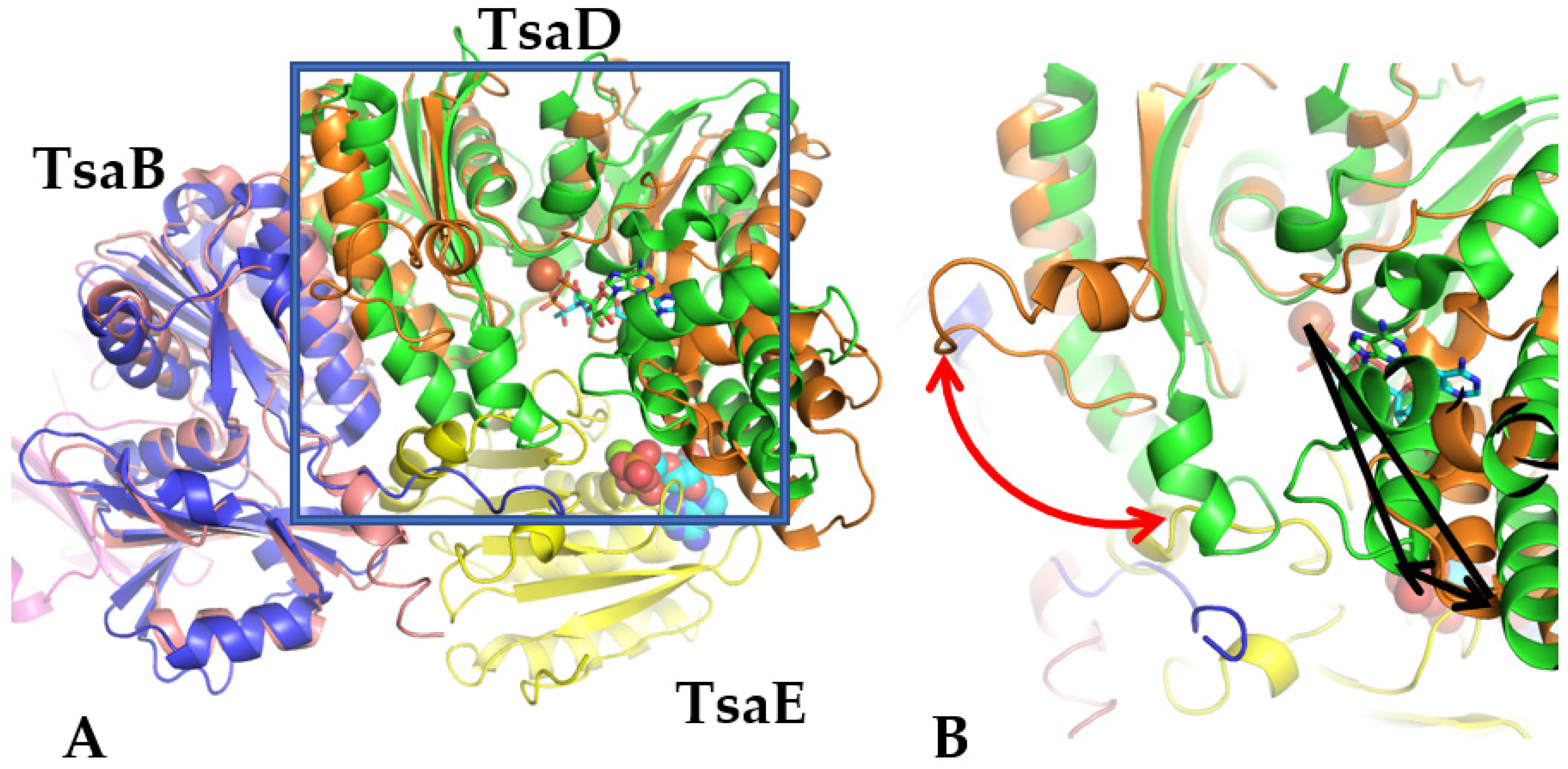
The bacterial TsaBD interface is highly similar to the interface in Qri7 (D-D homodimer), with a four-helical bundle formed by the helices from B and D components. The TsaBD heterodimer in
T. maritima followed the same logic at the BD interface but allowed for additional control by conformational changes in the TsaB element, particularly in the C-terminal helix, as seen in the comparison of 6N9A and 3ZEU [
11,
25]. This heterodimeric architecture creates an additional possibility for control of the complex through the binding of an additional element, TsaE. The control is accomplished by a significant conformational change in the region 30–50 of TsaD upon TsaE binding. The model of this fragment with the corresponding electron density is shown in
Figure S3. For instance, in the
T. maritima TsaBDE complex, upon binding of subunit E, there is an almost 90 degree rotation of this fragment away from subunit D, which is coupled with an opening of the active site cleft (around 17°) located at the junction of two domains of the D subunit (
Figure 2). Additionally, this change is accentuated by a transition from an extended to a helical conformation in the C-terminal fragment of TsaB. This complicated conformational transition is clearly illustrated by superposing the TsaBD heterodimer from
E. coli (4YDU) [
2] onto that in the
T. maritima TsaBD complex (
Figure 2).
The
E. coli and
B. subtilis complexes can be understood as half-complexes of
T. maritima. The
E. coli structure shows a much narrower cleft, at the apex of which the active site is located. Presumably, control of opening of this cleft is also needed for the binding of tRNA and transfer of the TC group to A37. As depicted in
Figure 2, the TsaE subunit clearly penetrates and clashes with the 30–40 fragment of the TsaD subunit in the
E. coli model, which results in the conformational transition mentioned above.
A similar change is observed in several KEOPS complexes. Binding of the Bud32 causes a significant rotation of the C-terminal helix, which protrudes into the space occupied by the postulated binding of tRNA. Therefore, the ATPase activity of Bud32 would exert a similar effect on the KEOPS complex in eukaryotes as the ATPase activity of TsaE in the BDE complex in bacteria [
11], despite a different fold and different location in the complex [
17].
3.3. Modeling of the tRNA Binding to the TsaD-like Subunits of the t6A37 Eukaryotic and Prokaryotic Complexes
The placement of TC-AMP at the active site of the TsaD component defined a specific location for the approaching A37 of the anticodon loop of tRNA (
Figure 3B and
Figure 4A). For the effective transfer of the threonylcarbamoyl group of TC-AMP to N6 of A37 to occur, the most likely position for N6 of the adenosine should be located within a 3.5 Å sphere around the TC-AMP scissile bond. Applying this constraint, we performed a comprehensive docking experiment utilizing all three representative complexes from mitochondria (Qri7), yeast (KEOPS), and
T. maritima (TsaBD 2:2 dimer) known to produce t
6A
37.
The docking experiments were carried out using the tRNA molecule from (1QF6) [
18], which was docked to the refined version of the complete Qri7 dimer, the DBBD dimer of the
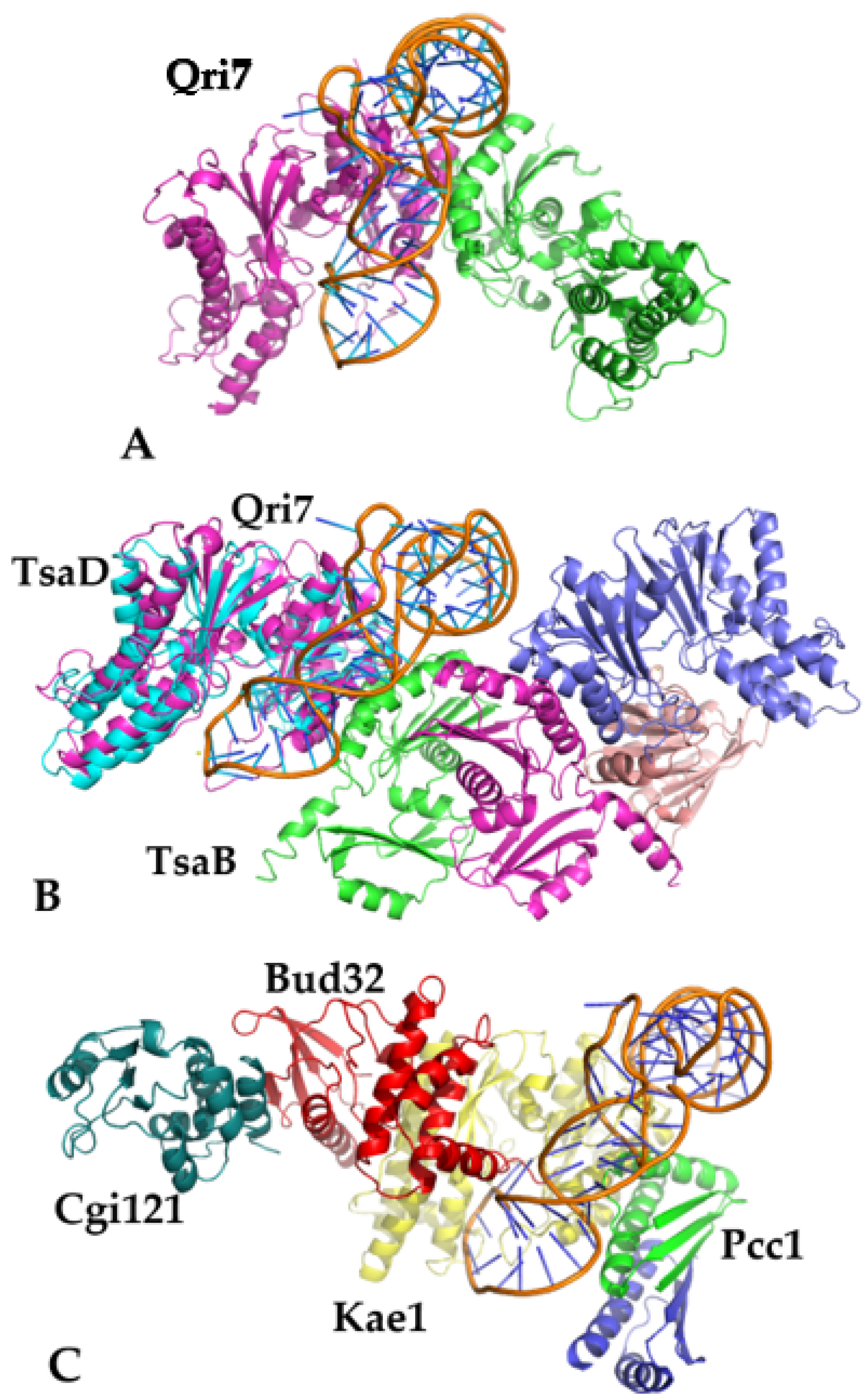
T. maritima complex described in this report (with TsaE omitted), and the partially reconstituted model of the yeast KEOPS complex of Kae1 with Pcc1. The full model of the yeast KEOPS complex with Kae1, Pcc1, Bud32, and Cgi121 was later used for visualization purposes and is shown in
Figure 4. The web service NPDock [
33] was used for docking experiments with the tRNA model that was extracted from PDB. The default settings of the program were used without post-refinement. The program produced 20,000 decoys/poses that were scored. Subsequently, the program clustered the 100 top solutions into 30 clusters. Only around 10 significantly populated clusters were identified within the limit of the 5 Å RMSD. The top four representative poses/clusters are shown in
Figure 3. All these poses, shown in
Figure 3A, allow for the N6 of A
37 to be positioned near the TC moiety and provide marginal but sufficient binding energy with the protein. Subsequently, more detailed criteria described below were applied, which indicated that cluster T1 was the most likely.
For selecting the best pose among the four dominant solutions (T1–4), the following rules were adopted: (1) the highest position on the pseudo-energy ranked list; (2) the best geometrical arrangement of A37 for the transfer of the TC moiety; and (3) the pose should have minimal interpenetration with volumes of auxiliary binding proteins (especially TsaC/C2 or TsaE) necessary for production and translocation of TC-AMP. Out of the four best clusters (T1–T4), only one satisfied all the conditions, which was T1. This pose has the most extensive protein-RNA contacts; it partially contacts the second molecule across the dimer interface, excluding the second molecule’s binding, and shows optimal capability for access to the active site of TsaD as the extended anticodon loop of the tRNA can easily be inserted into the active site (
Figure 3B). This approach would result in minimal distortion of the tRNA, similar to the one reported in 1QF6.
Out of three protein targets, the best score in pseudo-energy was obtained for Qri7, which lined up well with the expectation that Qri7 should be the best and minimal model for this interaction. The
T. maritima DBBD model produced intermediate results, while the least distinct solution was obtained for the Kae1 complex, as expected since this complex is the most regulated and serves multiple purposes. However, the best solution/poses for each protein-tRNA complex were in general agreement with each other, thus establishing a consensus binding pose that is utilized by the entire family, regardless of the presence of the auxiliary proteins (
Figure 4). This observation suggests a possible pathway for increasing regulatory control by the gradual addition of individual proteins to the catalytic core built around the TsaD subunit. This conclusion is supported by recent observations that the TsaE component in the TsaBDE complex [
4,
24], as well as Bud32 in the KEOPS complex [
5], bind independently and control tRNA binding by ATP hydrolysis at the site located at the interface between TsaD and TsaE or Bud32.
The universality of the binding pose for tRNA proposed in this report raises a very interesting question. Why are so many vastly different complexes involved in the same catalytic role of transferring the TC group to the A
37. The manner of tRNA binding described above can be regulated by the binding of auxiliary proteins. Alternative functions performed by these complexes provide an attractive hypothesis for explaining why such a variety of complexes exist. The eukaryotic KEOPS complex is known to possess DNA binding capabilities as well as telomere editing activity [
37]. This activity may require an increased ability to bind longer stretches of DNA, and such binding would be difficult to accomplish with a single Qri7-like dimer [
36]. The binding of auxiliary proteins not only provides control over the activity of the complex but apparently also increases binding. Therefore, the changes in composition of complexes contribute to the modulation of additional functions. New results show that the dimerization of the Kae1 complex through Pcc1 is not necessary for tRNA activity [
5], while this dimerization event may be required for DNA telomerase activity [
37].
Recently, two teams reported models of tRNA bound to complexes containing TsaD [
11,
38] derived from small-angle scattering experiments. These tentative models were constructed by fitting the tRNA molecules into the reported excess density extending from the fitted protein elements. In both cases, the incoming tRNA was interpreted as lying along the main axis of the complex. In TsaBD from
T. maritima [
11], the tRNA was fitted into the experimental density along the body of TsaD. A similar arrangement was detected in the full KEOPS complex, reinforced by the interpretation that the acetyl terminus of tRNA binds to Cgi121 [
38]. Guided by this observation, the authors of [
38] proposed that this pose represents the catalytically active complex of the tRNA with TsaD and extended this observation to all known TsaD representatives. The modeled complexes are presented in
Supplementary Figure S4h,i of [
38]. This model was reiterated by Beenstock & Schicheri [
39] in an excellent review paper that summarized the KEOPS results, as well as in the latest review [
1]. The pose proposed for KEOPS was applied to other TsaD representatives, despite the fact that in Qri7 and other TsaBD complexes, the interface would be smaller and apparently insufficient to form a stable complex. The same paper stated that Cgi121 is expressed in sub-stoichiometric ratios to the remaining components of the complex, thus undermining the conclusion that Cgi121 participates in the catalytically active tRNA complex. Additionally, van Tilbeurgh [
13], through studying the association of Bud32 with Cgi121, concluded that Cgi121 cannot participate in the active complex as its deletion does not inhibit the synthesis of t
6A modification [
12,
39,
40].
In both complexes, this conformation of the tRNA would make the approach to the active site very difficult with the anticodon loop at the productive conformation, as suggested by [
6]. Additionally, this pose would interfere with the regulatory roles of TsaE or Bud32. Therefore, one can conclude that both experimental results, which are based on small-angle scattering, can be interpreted as a transient binding that is detected in its early stages and that conforms to one of the four solutions described above for the tRNA bound to the TsaD (pose T3). Particularly, binding of tRNA to Cgi121 should be treated as an initial contact phenomenon in which Cgi121 fulfills the role of the tRNA chaperone to bring it up close to the large KEOPS complex. However, it is less mobile than its bacterial counterparts to effectively perform the assigned function. These arguments are reinforced by noticing that the KEOPS complex performs the catalysis with the telomeres as a substrate, which are larger in size than a single tRNA molecule.
3.4. Modeling of the Catalytic Reaction of TsaD
To facilitate the formulation of the proposal for the catalytic reaction carried out by the TsaD, we utilized the 3-D model of the substrates bound to its active site. The refinement suggested a location of the TC-AMP (6NAK) that was confirmed later by the structure 6Z81, as well as the placement of the A37 by docking of the tRNA. The corrected and refined model of a BDE dimer (6NAK, derived from 6FPE) was used for docking of tRNA with the threonycarbamoyl modification (TC) removed (1QF6). Before docking, the E element was removed from the model, and in one of the D subunits, the region 30–50 was modeled in a conformation corresponding to the structure of the BD dimer from bacteria (4YDU) that attains a more extended conformation. The manually adjusted pose/decoy number 13,497 was used as the representative of the tRNA binding mode to the TsaD. Such a model was used for modeling the catalytic reaction and is presented in
Figure 5 and
Figure S6.
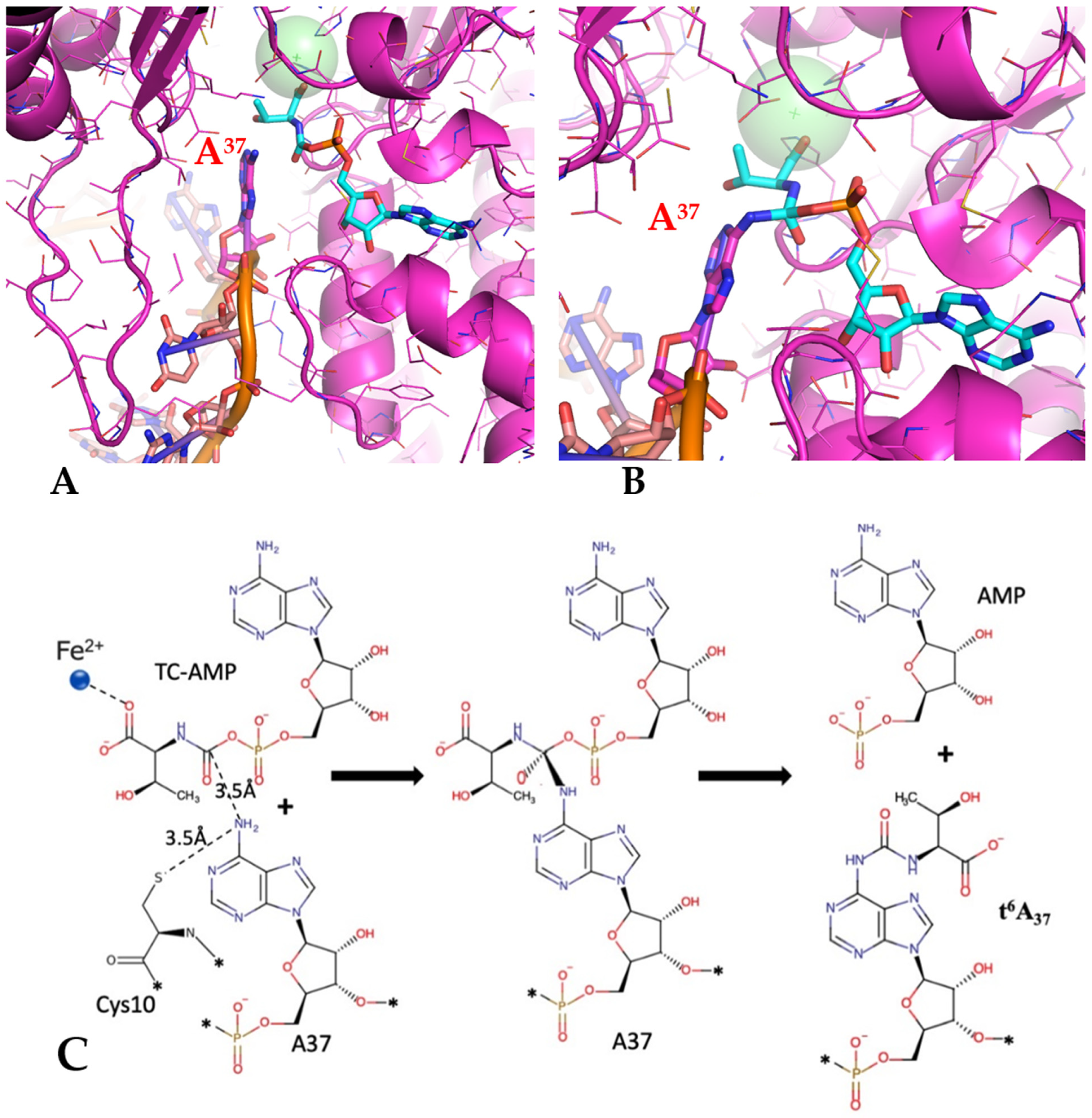
The location and orientation of the TC-AMP in the active site of the TsaD component allow for formulating a proposal for the catalytic mechanism carried out by TsaD. Such a mechanism is further supported by the structural results of the TC-AMP analog bound to the
E. coli TsaBD complex (6Z81) [
23]. The mechanism of transfer of the threonylcarbamoyl moiety can be postulated as a nucleophilic attack of the activated N6 nitrogen of adenosine on the bridging carbamate group of TC-AMP, followed by the formation of an unstable tetrahedral intermediate (
Figure 5A,B). This spatial arrangement takes advantage of a partial resonant structure of the carbamate, reminiscent of a peptide amide bond in peptides, that allows for the proteolytic activities of different agents/nucleophiles in several classes of proteases. The unstable tetrahedral intermediate would later collapse to the final product, driven by AMP as a leaving group. Therefore, the location of the flat face of the carbamate group in TC-AMP defines the location of the approaching N6 of the adenosine at the side directionality with sufficient accuracy for modeling (
Figure 3B and
Figure 5A). The position for a deprotonated N6 is defined by the face-to-face contact of the adenosine ring at position 37 and the amide bond plane of TC-AMP, which is bracketed on the opposite site by a loop containing Cys10 in
T. maritima.
The only remaining issue, besides a precise location of the adenosine ring determined by the tRNA directional approach, which is controlled by the three-dimensional organization of the active site cleft in TsaD, is the mechanism of activation/deprotonation at N6 of A37. As is well remembered from the early days of DNA modeling, the main contribution to the discovery of DNA structure by Watson and Crick was the fact that the bases can be present in solution in different tautomeric states [
41,
42]. Therefore, full and controlled deprotonation of adenosine at N6 is not needed for the reaction to proceed. Complete deprotonation is heavily disadvantaged by a high pKa value of N6. Instead, an internal proton migration between N6 and N1 to form a different tautomeric state would be preferred. Therefore, only a partial stabilization of one of the presented states is needed for the reaction to proceed without an elaborate deprotonation mechanism required by acid-base reactions. The best candidate for such stabilization in
T. maritima is a member of the highly conserved sequence SCDE, i.e., Cys10 of TsaD, which in the crystal structure (6NAK) is located with its Sγ group around 7 Å away from the flat face of the scissile bond in TC-AMP, therefore in close proximity to the inserting N6. The size of the active site cleft is perfectly suitable for insertion of the individual base, and it is compatible with helical separation of bases in nucleic acids, therefore providing direct van der Waals contacts. The protonation state of Cysteine residue is most likely controlled by the closeness of the charged residues at the loop (SCDE) and an additional contact with Asp296 that participates directly in the binding of the metal ion at the active site of TsaD. The model of the transition state of the proposed reaction is presented in
Figure 5B, and the postulated reaction’s schematic is presented in
Figure 5C.
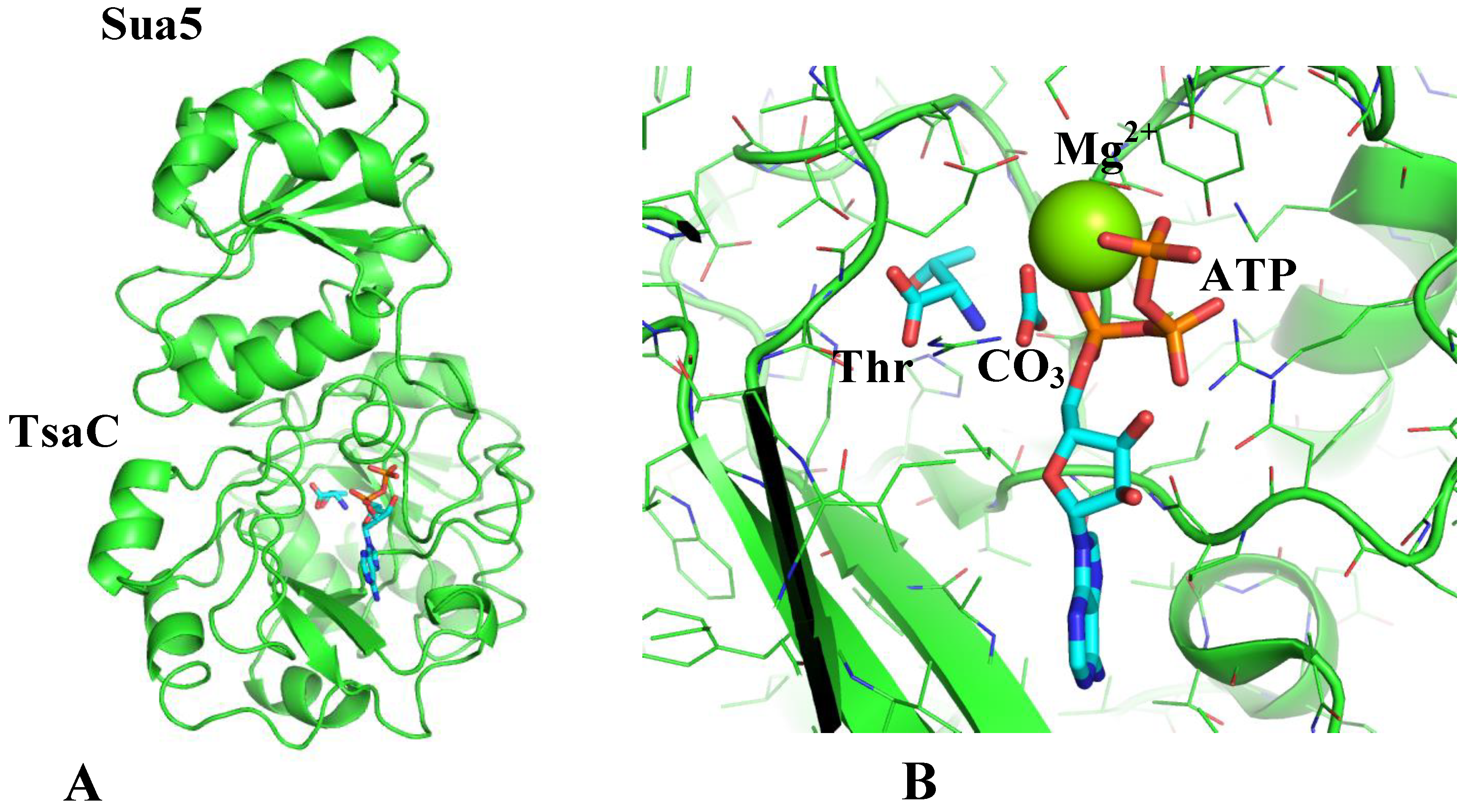
Figure 3B shows the model of a ternary binding complex of TsaC2 (threonylcarbamoyl synthase) and tRNA to TsaD. This model shows a tentative location of the Sua5 domain, which is highly mobile, as was suggested by small-angle scattering studies of the mobility of TsaC2 from
B. burgdorferi [
43]. Even though the model places tRNA and TsaC on opposite sides of the TsaD subunit of the complex, their binding must be transient and anti-cooperative, as all experimental attempts to capture these complexes have so far been unsuccessful. Additionally, binding of TsaE must be antagonistic to binding of both tRNA and TsaC, as suggested by experimental results [
10] and inferred from overlap of different components when these are modeled bound to TsaD simultaneously.
3.5. Modeling of the Catalytic Reaction of TsaC
The mutual orientation of the TsaC versus the TsaD can be well modeled (
Figure 3) in analogy to the domain organization of the structure of TobZ. This model suggests a clear path of transfer for unstable TC-AMP between both active sites that are separated by around 15 Å in TobZ as well as in our model. It is well documented that unstable intermediates are protected at the active sites of enzymes in internal protein cavities [
44,
45] and can survive for much longer times than their natural half-lifetime in solution. A short distance and similar substrates/product directionality in the active sites between TsaC and TsaD offer a necessary condition for an effective transfer. An effective proxy for the general binding affinity to both enzymes’ active sites can be the number of hydrogen bonds formed to the adenosine ring. The increased number of hydrogen bonds to the substrate by TsaD as compared to TsaC, combined with a short distance to diffuse a synthesized unstable intermediate, TC-AMP, provides a feasible driving force for a rapid transfer and for the stabilization of TC-AMP.
The structures and the mechanism of TsaC have been extensively investigated. Despite this research effort, the catalytic mechanism remains unclear, and the main question of what is the preferred substrate, CO2 or HCO3, has not been decisively resolved. After an extensive review of the existing literature, we decided to produce a 3-D model of the binding of substrates to shed light on the steric and electronic requirements imposed by the TsaC active site. The comparison between the details of different structures indicated several important features.
The first is that TsaC structures lack a distinct and specific metal ion binding site that would drive and organize the sequential binding of substrates. Metal ions are detected only in structures with bound nucleotides. This fact can be attributed to the necessity of charge neutralization of nucleotide triphosphates in solution. The presence of metal counterions bound to the nucleotides suggests that upon binding of the nucleotides, the metal ion would be brought to the active site of the particular protein with them. In models of TsaC/C2 structures with bound metal ions such as T. maritima, P. abbyssi, and S. tokodaii, the only residue that directly coordinates the metal ion is Ser, which explains its rather weak affinity to the apo-protein.
The second important feature is that there is a well-defined and conserved location for L-Thr, which stands in contrast to variations/shifts in the positions of the phosphates of nucleotides in different structures. The structure containing the L-Thr and the AMPPNP shows a significant shift of the alpha phosphate compared to that containing the product TC-AMP. This feature suggests that binding of L-Thr provides a structural base for binding of the second ligand (CO2 or CO3), which in turn facilitates binding of the ATP with the associated metal ion. Most likely, the incoming metal ion (Mg2+) binds not only the triphosphate but also the second ligand, CO2 or CO3.
The lack of a well-defined metal ion binding site clearly projects onto the problem of selecting which of the substrates is preferred by TsaC: CO
2 or CO
3. The enzymes utilizing the gaseous CO
2, such as RUBISCO or carbonic anhydrase (CA), provide restrictive conditions for the binding of this molecule. As the presence of dissolved CO
2 dramatically drops with increased pH, in both enzymes there is a strongly bound metal ion to organize their active sites, and additionally, in vivo, the cells utilize an additional mechanism for concentrating the CO
2. Both methods must be used by the enzymes, as the natural solubility and availability of CO
2 would limit the catalytic turnover. Clearly, the TsaC enzymes do not have any of these requirements fulfilled. Even in enzymes that were suggested to use CO
2, such as biotin carboxylase, the use of metal ions and transient CO
3 intermediates that produce carboxyphosphate [
46] suggests that CO
3 is a much more abundant and dominant species.
Considering all these factors and the relative availability of CO
2 or CO
3, it becomes natural to consider the possibility that HCO
3 is a preferred substrate for the TsaC/C2. Additional experimental support for this choice is provided by the structure of
Pyrococcus abyssi Sua5 (6F89), in which, for the first time, CO
3 was tentatively captured [
19]. However, despite our clear preference for CO
3 as a ligand, we need to consider a model for CO
2 initiating the reaction. To provide an equal basis to compare the advantages and drawbacks of the particular scheme we sketched out below, both schemes are shown in
Figure 6. Based on all these observations, we proposed the catalytic mechanism for the TsaC-like enzymes (threonylcarbamoylate synthase) and constructed the 3-D model of its initial state and subsequent intermediates (
Figure 6) for both alternative mechanisms.
The proposal for the mechanism was aided by the 3-D model of the substrates bound to TsaC2. The model of the TsaC2 with all three substrates at the active site was obtained in the following manner. The model was generated from the model of TsaC2 with ATP and Thr (3AJE). This model was compared/superposed with the model of the TsaC2 with the product of the reaction TC-AMP (4E1B). This comparison revealed the conformational/rotational change/movement of the alpha phosphate of the ATP. Modeling this motion created room for the third substrate, CO
3, sandwiched between the Thr and the alpha phosphate of the ATP. In placing the CO
3, we were guided by the model (6Z81). The final model was created by adjusting the initial locations of CO
3 and ATP for optimal contacts and postulating the location of the metal in the active site. The resulting model is shown in
Figure 6B and in
Figure S7.
Both mechanisms require the sequential binding of the substrates: first, L-Thr; second, CO
2 or CO
3; and last, adenosine triphosphate (ATP). In our preferred mechanism, where the substrate is CO
3, the reaction must proceed through the condensation of three sequentially bound substrates: L-threonine, CO
3, and ATP (
Figure 6B) and would produce (TC-AMP), diphosphate, and a water molecule. For details of the reaction, please refer to the schematic presented in
Figure 6C. In the first step, the activated/deprotonated form of carbonate performs a nucleophilic attack with the O
− group on the alpha phosphate of adenosine, and by inversion of configuration, it produces carboxy-AMP and diphosphate as a leaving group. This happens as soon as the adenosine triphosphate binds and brings the metal ion to the active site, where it coordinates the carbonate ion and the phosphate group of ATP. Incidentally, highly unstable carboxy-AMP was tentatively captured in structure 6N9A [
11]. Carboxy-AMP is unstable because it is prone to a nucleophilic attack by the water molecule, so in an aqueous environment, it would undergo excision of the carboxyl group in reaction with the water molecule, thus re-creating carbonate. In the active site of the enzyme, in the absence of an activated water molecule, the deprotonated amine group (NH
2) of Thr performs the nucleophilic attack, and the collapsing tetrahedral intermediate produces the product threonylcarbamoylate-AMP (TC-AMP) with the release of a water molecule. This step is very reminiscent of a standard Shiff base mechanism in which a proton is transferred from the amine group to the leaving oxygen, thus releasing a water molecule. The unstable product TC-AMP is subsequently transferred to the active site of TsaD, which performs the final step of the t
6A
37 pathway, as described before.
If one assumes that the preferred ligand is CO2, then the only choice for the initial step would be a nucleophilic attack of the amine group of Thr to create the carbamyl group at the terminal nitrogen. In a subsequent step, this carbamate would have to attack the alpha phosphate of ATP to produce the threonylcarbamoylate-AMP (TC-AMP) with a concomitant release of a diphosphate.
Both choices for the reaction have ample support in the literature. However, our modeling of the 3-D structures strongly supports the view that a preferred ligand is CO
3. There is no apparent chemical signature for CO
2 binding to the TsaC/C2 active site; therefore, there is no apparent activation mechanism for CO
2 activation to complete the carbamate formation. Additionally, CO
2 abundance at pH ~7.5, which is routinely utilized in kinetic experiments and probably prevails at physiological conditions, would indicate that its concentration is below 1%, as judged by the Bjerrum plot. Hence the dominant form would be CO
3 at this pH value, with a 99:1 ratio. Finally, there are many examples of CO
3 interactions with phosphates and the production of unstable intermediates. The best examples are provided by the structures and biochemistry of carbamoyl-phosphate synthase [
46,
47]. Nevertheless, to resolve the problem of preferred ligands, careful biochemical experiments mapping the entire pH profile for such a reaction should be performed.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






