
Axial Tomography in Live Cell Microscopy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- -
- embedding of the cells in an appropriate medium within rotatable devices as well as precise adjustment of various angles under the microscope [41];
- -
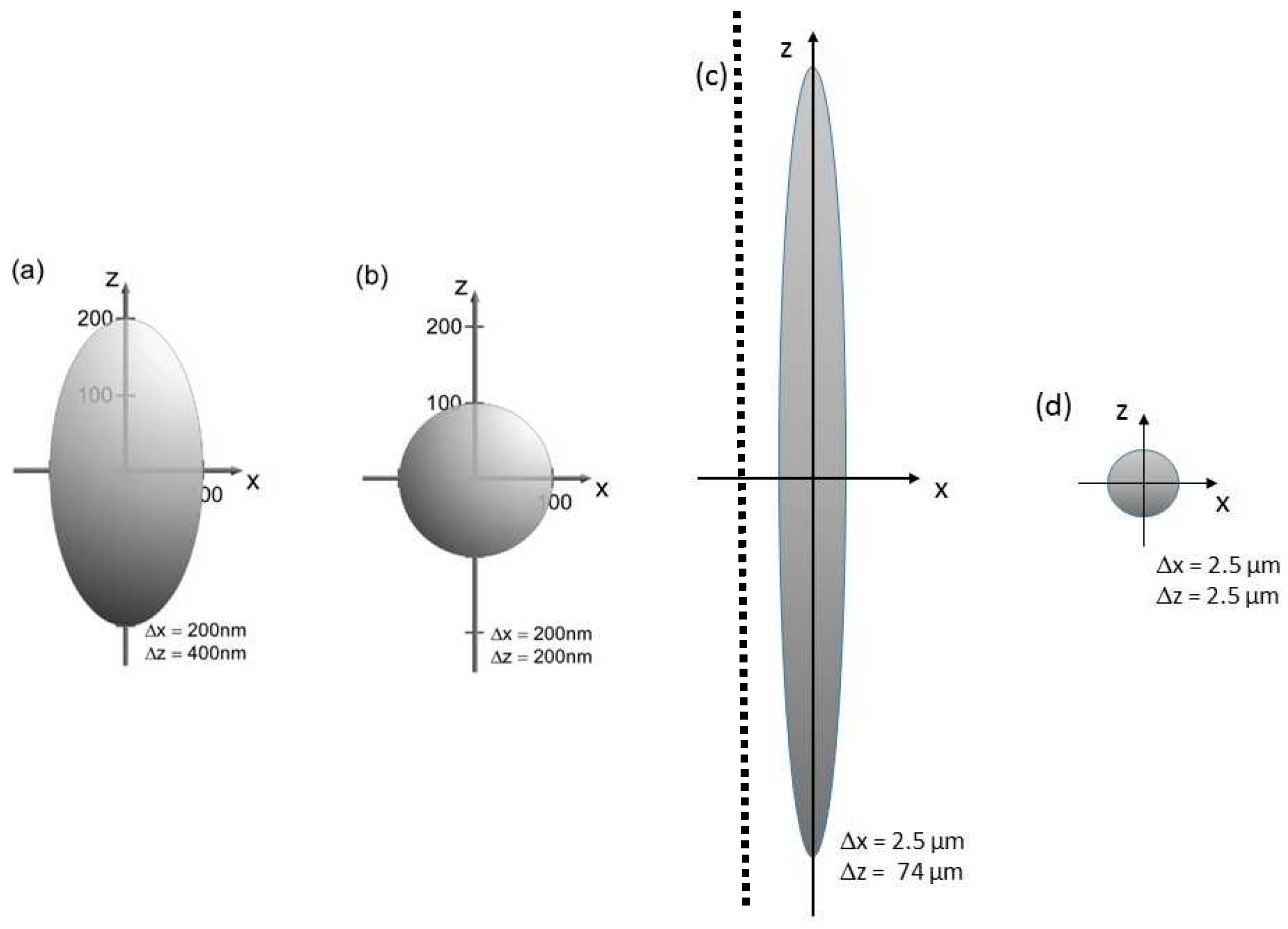
2. Angular Resolution
3. Axial Tomography in 3D Microscopy
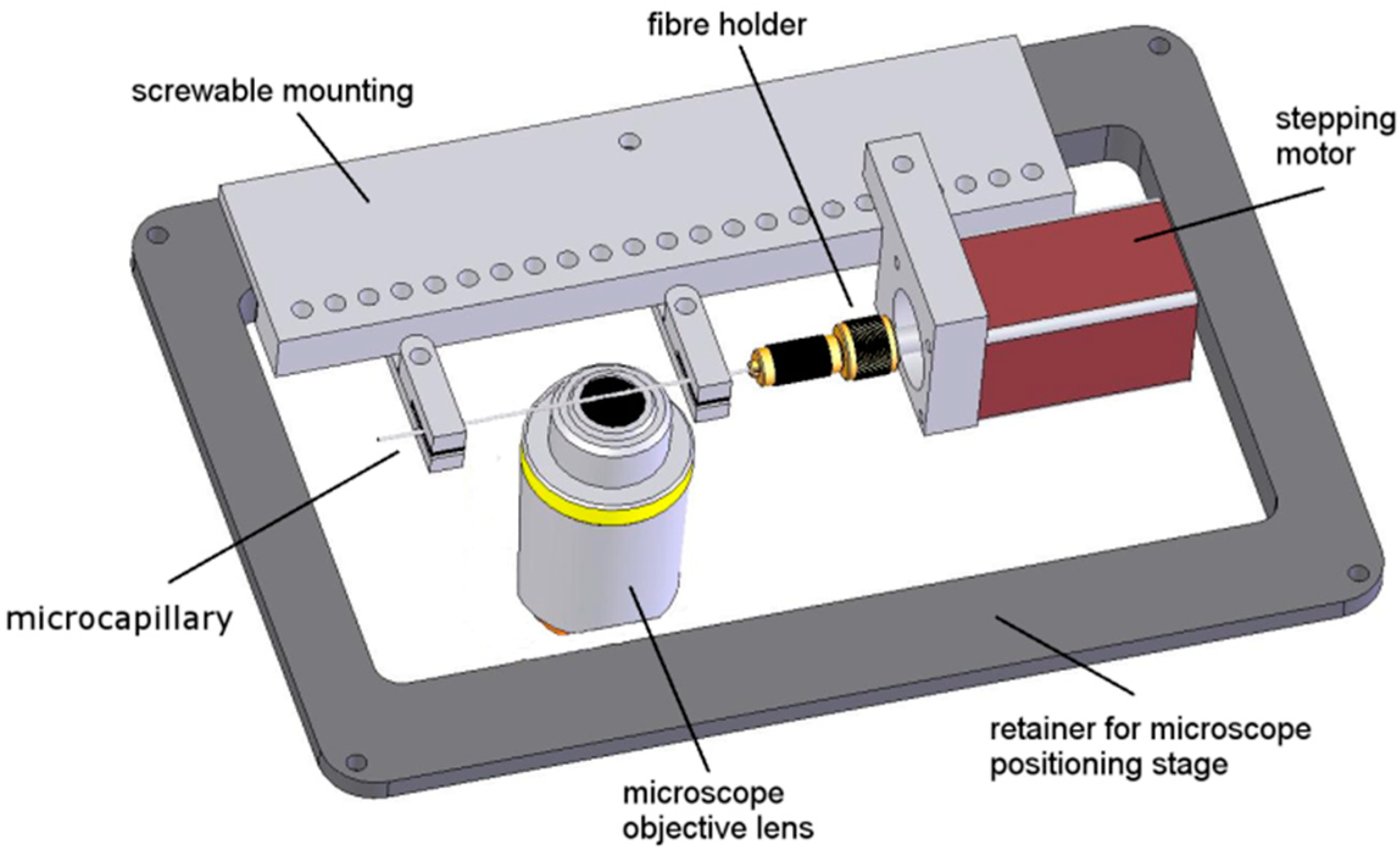
3.1. Overview
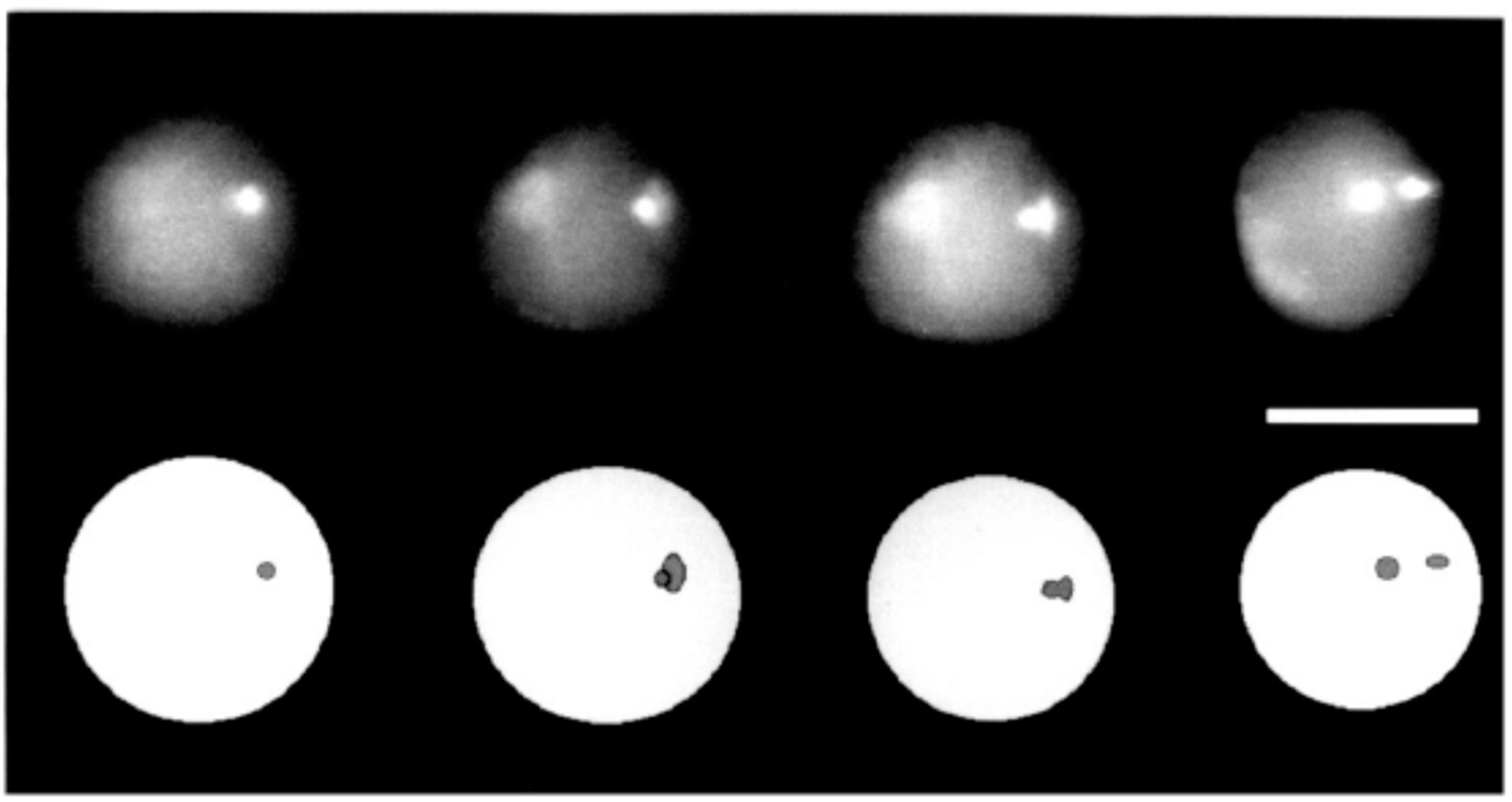
3.2. 3D Distance Measurements at the Nanoscale
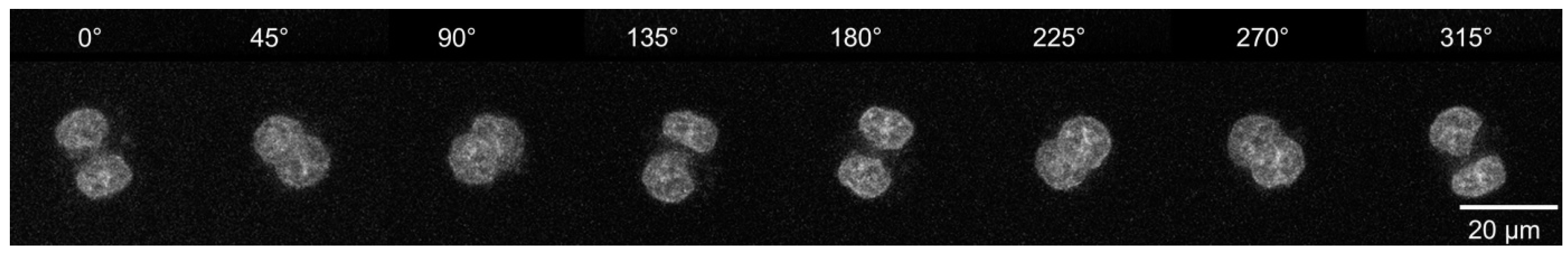
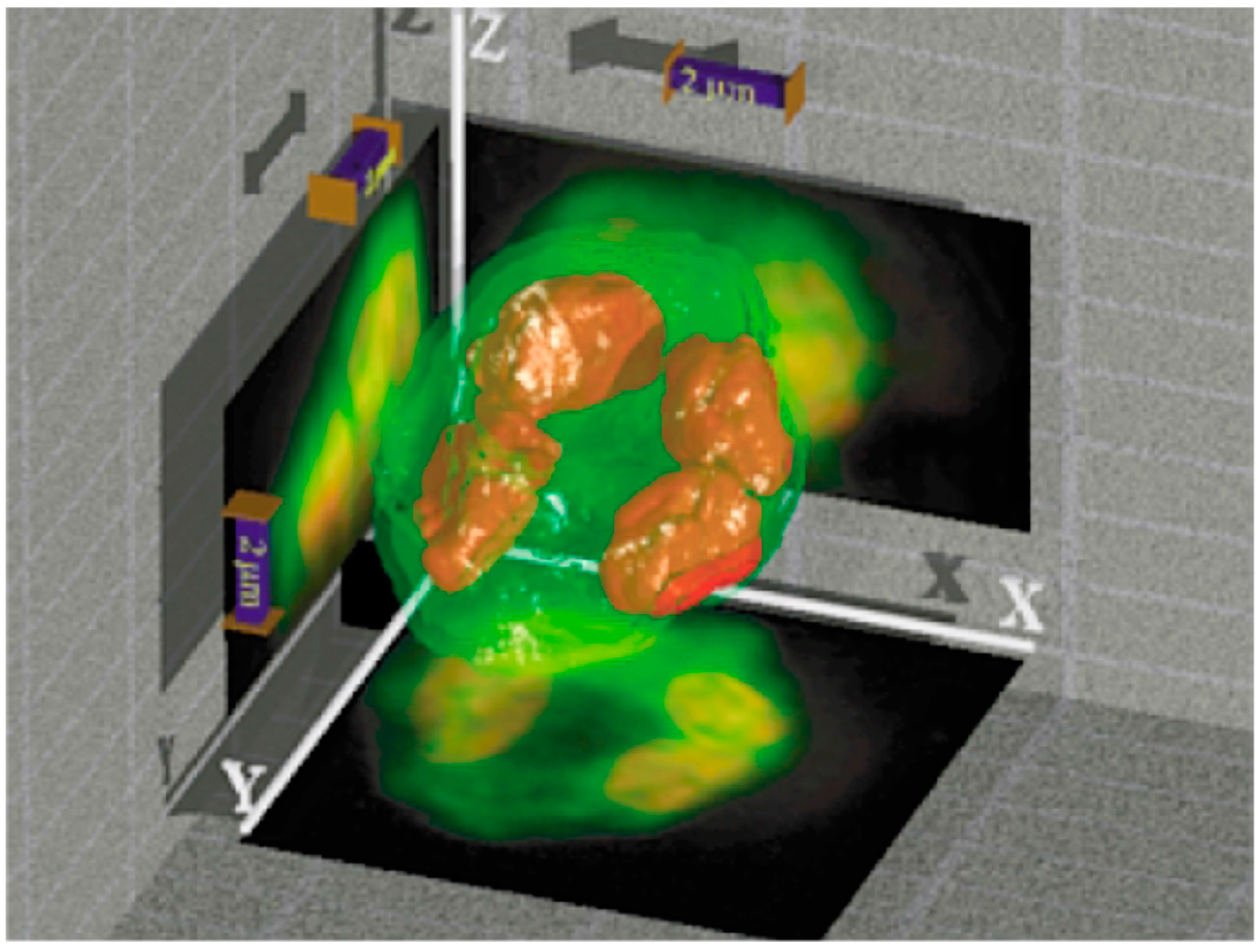
3.3. Axial Tomography to Study the 3D Distribution of Selected Chromatin Sites
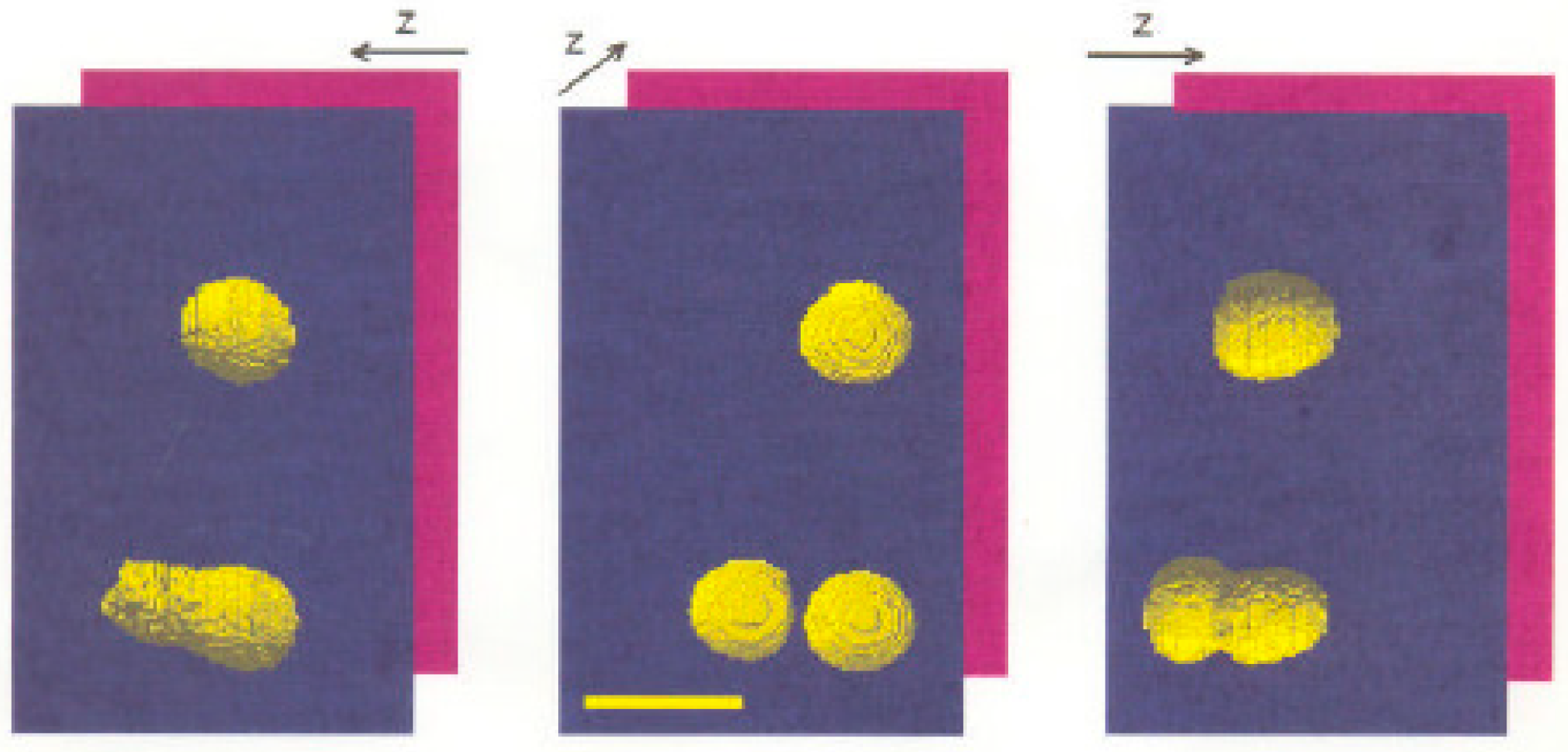
3.4. Computational Reconstruction of 3D Images from Axial Tomography Data
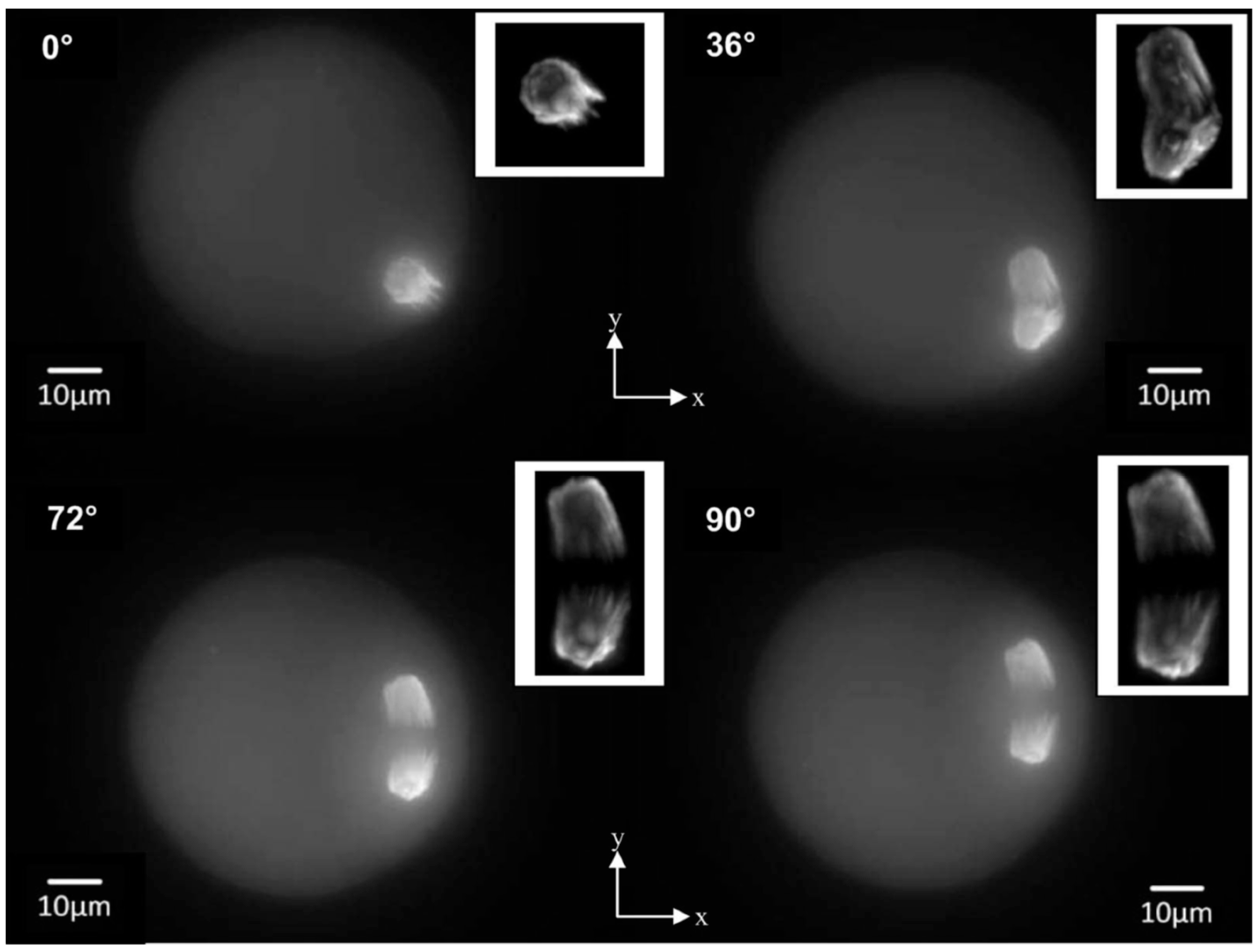
3.5. Application of Axial Tomography in Embryology
3.6. Combination of Axial Tomography and Structured Illumination (SIM)
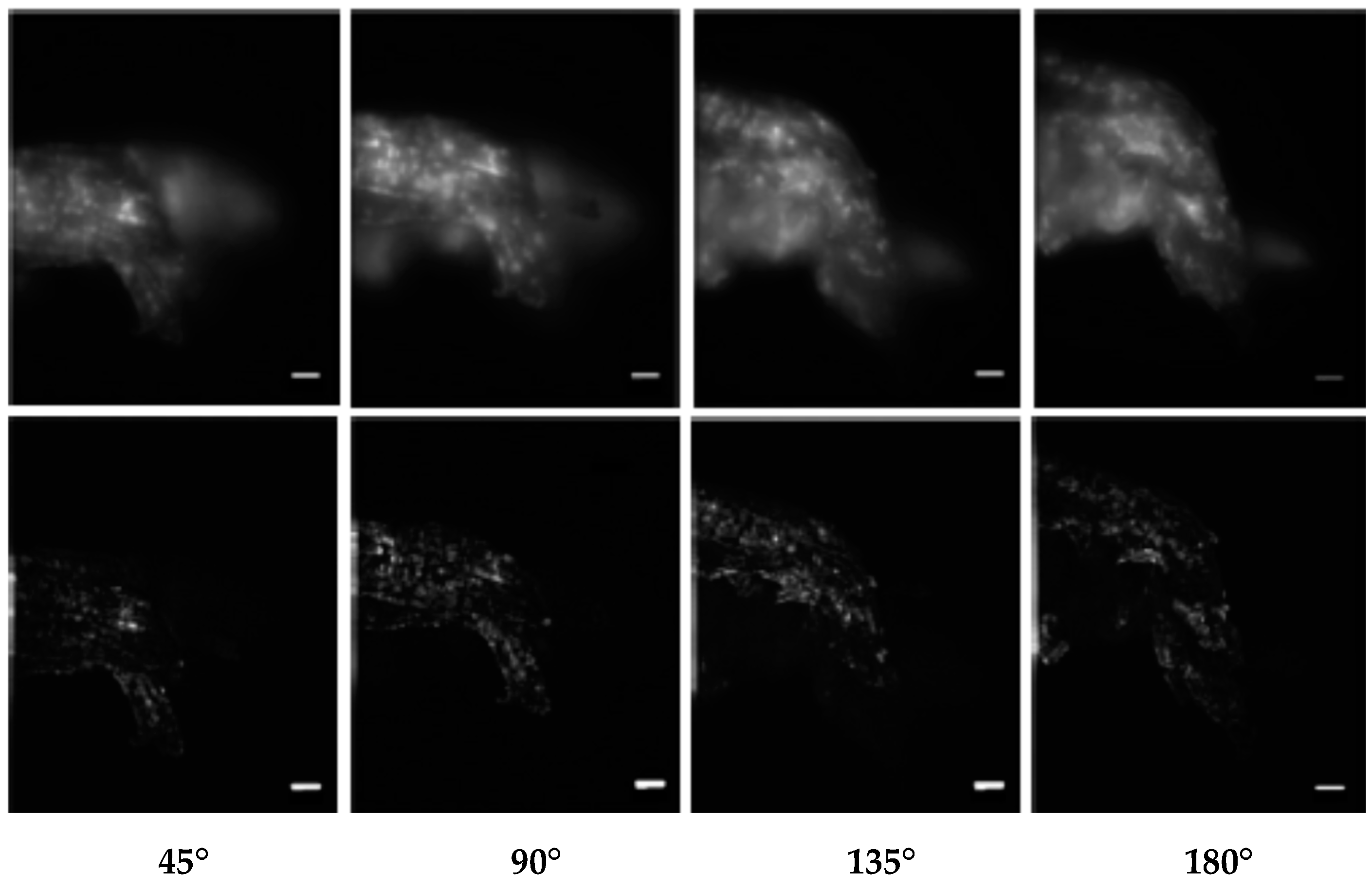
3.7. Axial Tomography of Very Large Transparent Objects: Combination with Ring-Array Microscopy
4. Discussion
4.1. Simplicity
4.2. Multiple Measurements of the Same Object
4.3. Deep View Imaging of Very Large Objects
4.4. Molecular Imaging/FRET
4.5. Final Remarks Concerning 3D Reconstruction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Betzig, E. Proposed method for molecular optical imaging. Opt. Lett. 1995, 20, 237–239. [Google Scholar] [CrossRef]
- Hess, S.; Girirajan, T.; Mason, M.D. Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Geisler, C.; Wurm, C.A.; von Middendorff, C.; Jakobs, S.; Schoenle, A.; Egner, A.; Hell, S.W.; Eggeling, C. Two-color far-field fluorescence nanoscopy based on photoswitchable emitters. Appl. Phys. B 2007, 88, 161–165. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Sub-diffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008, 47, 6172–6176. [Google Scholar] [CrossRef] [PubMed]
- Biteen, J.S.; Thompson, M.A.; Tselentis, N.K.; Bowman, G.R.; Shapiro, L.; Moerner, W.E. Single-molecule active-control microscopy (SMACM) with photo-reactivable EYFP for imaging biophysical processes in live cells. Nat. Methods 2008, 5, 947–949. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; E Jones, G.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2011, 9, 195–200. [Google Scholar] [CrossRef]
- Lidke, K.A.; Rieger, B.; Jovin, T.M.; Heintzmann, R. Super-resolution by localization of quantum dots using blinking statistics. Opt. Express 2005, 13, 7052–7062. [Google Scholar] [CrossRef]
- Cremer, C.; Masters, B.R. Resolution enhancement techniques in microscopy. Eur. Phys. J. 2013, 38, 281–344. [Google Scholar] [CrossRef]
- Cremer, C.; Szczurek, A.; Schock, F.; Gourram, A.; Birk, U. Super-resolution microscopy approaches to nuclear nanostructure imaging. Methods 2017, 123, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffie’, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Primers 2021, 1, 39. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.C.M.; Masullo, L.A.; Baudrexel, I.; Steen, P.R.; Kowalewski, R.; Eklund, A.S.; Strauss, S.; Unterauer, E.M.; Schlichthaerle, T.; Strauss, M.T.; et al. Ångström-resolution fluorescence microscopy. Nature 2023, 617, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef]
- Wildanger, D.; Medda, R.; Kastrup, L.; Hell, S. A compact STED microscope providing 3D nanoscale resolution. J. Microsc. 2009, 236, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Far-Field Optical Nanoscopy. Science 2007, 316, 1153–1158. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef]
- Liu, S.; Hoess, P.; Ries, J. Super-Resolution Microscopy for Structural Cell Biology. Annu. Rev. Biophys. 2022, 51, 301–326. [Google Scholar] [CrossRef]
- Lukosz, W. Optical systems with resolving powers exceeding the classical limit, Part 1. J. Opt. Soc. Am. 1966, 56, 1463–1472. [Google Scholar] [CrossRef]
- Lukosz, W. Optical systems with resolving powers exceeding the classical limit, Part II. J. Opt. Soc. Am. 1967, 57, 932–941. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L. Surpassing the Lateral Resolution Limit by a Factor of Two Using Structured Illumination Microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. Proc. SPIE 1999, 356, 185–196. [Google Scholar]
- Gustafsson, M.G.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-Dimensional Resolution Doubling in Wide-Field Fluorescence Microscopy by Structured Illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, L.M.; Wicker, K.; Mandula, O.; Heintzmann, R. Structured illumination microscopy of a living cell. Eur. Biophys. J. 2009, 38, 807–812. [Google Scholar] [CrossRef]
- Bailey, B.; Farkas, D.; Taylor, D.L.; Lanni, F. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature 1993, 366, 44–48. [Google Scholar] [CrossRef]
- Lanni, F.; Bailey, B.; Farkas, D.; Taylor, D.L. Excitation field synthesis as a means for obtaining enhanced axial resolution in fluorescence microscopes. Bioimaging 1993, 1, 187–196. [Google Scholar] [CrossRef]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905–1907. [Google Scholar] [CrossRef]
- Cremer, C.; Birk, U. Spatially modulated illumination microscopy: Application perspectives in nuclear nanostructure analysis. Philos. Trans. R. Soc. A 2022, 380, 20210152. [Google Scholar] [CrossRef]
- Birk, U. Super-Resolution Microscopy—A Practical Guide; Wiley-VCH: Weinheim, Germany, 2017; 408p, ISBN 978-3-527-34133-7. [Google Scholar]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer: Boston, MA, USA, 2006. [Google Scholar] [CrossRef]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996, 59, 427–471. [Google Scholar] [CrossRef]
- Huisken, J.; Swoger, J.; del Bene, F.; Wittbrodt, J.; Stelzer, E.H.K. Optical sectioning deep inside live embryos by SPIM. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef]
- Pampaloni, F.; Chang, B.-J.; Stelzer, E.H. Light sheet-based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissues. Cell Tissue Res. 2015, 360, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Santi, P.A. Light Sheet Fluorescence Microscopy. J. Histochem. Cytochem. 2011, 59, 129–138. [Google Scholar] [CrossRef]
- Hou, W.; Wei, Y. Evaluating the resolution of conventional optical microscopes through point spread function measurement. iScience 2023, 26, 107976. [Google Scholar] [CrossRef]
- Booth, M. Adaptive Optics for Microscopy, In Microscope Resolution Estimation and Normalised Coordinates (1.0). 2020. [CrossRef]
- Hell, S.; Stelzer, E.H.K. Properties of a 4Pi confocal fluorescence microscope. J. Opt. Soc. Am. 1992, 9, 2159–2166. [Google Scholar] [CrossRef]
- Hänninen, P.E.; Hell, S.W.; Salo, J.; Soini, E.; Cremer, C. Two-photon excitation 4Pi confocal microscope: Enhanced axial resolution microscope for biological research. Appl. Phys. Lett. 1995, 66, 1698–1700. [Google Scholar] [CrossRef]
- Bahlmann, K.; Jakobs, S.; Hell, S.W. 4Pi-confocal microscopy of live cells. Ultramicroscopy 2001, 87, 155–164. [Google Scholar] [CrossRef]
- Bruns, T.; Schickinger, S.; Schneckenburger, H. Single plane illumination module and micro-capillary approach for a wide-field microscope. J. Vis. Exp. 2014, 90, e51993. [Google Scholar] [CrossRef]
- Heintzmann, R.; Cremer, C. Axial tomographic confocal fluorescence microscopy. J. Microsc. 2002, 206, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Saalfeld, S.; Schindelin, J.; Tomancak, P. Software for bead-based registration of selective plane illumination microscopy data. Nat. Methods 2010, 7, 418–419. [Google Scholar] [CrossRef]
- Landis, W.J.; Song, M.J.; Leith, A.; McEwen, L.; McEwen, B.F. Mineral and organic matrix interaction in normally calcifying tendon visualized in three dimensions by high-voltage electron microscopic tomography and graphic image reconstruction. J. Struct. Biol. 1993, 110, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Bradl, J.; Hausmann, M.; Ehemann, V.; Komitowski, D.; Cremer, C. A tilting device for three-dimensional microscopy: Application to in situ imaging of interphase cell nuclei. J. Microsc. 1992, 168, 47–57. [Google Scholar] [CrossRef]
- Gage, S.H. Special oil immersion objectives for dark-field microscopy. Science 1921, 54, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, W.J.; Wilson, J.D.; Foster, T.H. Microscope enabling multimodality imaging, angle-resolved scattering, and scattering spectroscopy. Opt. Lett. 2007, 32, 2348–2350. [Google Scholar] [CrossRef] [PubMed]
- Rothe, T.; Schmitz, M.; Kienle, A. Angular and spectrally resolved investigation of single particles by darkfield scattering microscopy. J. Biomed. Opt. 2012, 17, 117006. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Voit, F.; Kienle, A.; Schneckenburger, H. Light scattering microscopy with angular resolution and its possible application to apoptosis. J. Microsc. 2015, 257, 1–7. [Google Scholar] [CrossRef]
- Thompson, N.L.; Burghardt, T.P.; Axelrod, D. Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys. J. 1981, 33, 435–454. [Google Scholar] [CrossRef]
- Burmeister, J.S.; Truskey, G.A.; Reichert, W.M. Quantitative analysis of variable-angle total internal reflection fluorescence microscopy (VA-TIRFM) of cell/substrate contacts. J. Microsc. 1994, 173 Pt 1, 39–51. [Google Scholar] [CrossRef]
- Axelrod, D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 1981, 89, 141–145. [Google Scholar] [CrossRef]
- Sund, S.E.; Axelrod, D. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching. Biophys. J. 2000, 79, 1655–1669. [Google Scholar] [CrossRef]
- Betz, W.J.; Mao, F.; Smith, C.B. Imaging exocytosis and endocytosis. Curr. Opin. Neurobiol. 1996, 6, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Oheim, M.; Loerke, D.; Stühmer, W.; Chow, R.H. The last few milliseconds in the life of a secretory granule. Eur. J. Biophys. 1998, 27, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Sailer, R.; Strauss, W.S.; Lyttek, M.; Steiner, R.; Schneckenburger, H. Variable-angle total internal reflection fluorescence microscopy (VA-TIRFM): Realization and application of a compact illumination device. J. Microsc. 2003, 211 Pt 1, 19–29. [Google Scholar] [CrossRef]
- Stout, A.L.; Axelrod, D. Evanescent field excitation of fluorescence by epi-illumination microscopy. Appl. Opt. 1989, 28, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, M.; Hérault, K.; Oheim, M. Eliminating unwanted far-field excitation in objective-type TIRF. Part II: Combined evanescent-wave excitation and supercritical-angle fluorescence detection improves optical sectioning. Biophys. J. 2014, 106, 1044–1056. [Google Scholar] [CrossRef]
- Verdes, D.; Ruckstuhl, T.; Seeger, S. Parallel two-channel near- and far-field fluorescence microscopy. J. Biomed. Opt. 2007, 12, 034012. [Google Scholar] [CrossRef] [PubMed]
- Ter-Pogossian, M.M. Basic Principles of Computed Axial Tomography. Semin. Nucl. Med. 1977, 7, 109–127. [Google Scholar] [CrossRef]
- Duliu, O.G. Computer Axial Tomography in Geosciences: An Overview. Earth Sci. Rev. 1999, 48, 265–281. [Google Scholar] [CrossRef]
- Bradl, J.; Rinke, B.; Schneider, B.; Edelmann, P.; Krieger, H.; Hausmann, M.; Cremer, C. Resolution improvement in 3D fluorescence microscopy by object tilting. Eur. Microsc. Anal. 1996, 44, 9–11. [Google Scholar]
- Matula, P.; Kozubek, M.; Staier, F.; Hausmann, M. Precise 3D image alignment in micro-axial tomography. J. Microsc. 2003, 209, 126–142. [Google Scholar] [CrossRef]
- Miao, Q.; Rahn, J.R.; Tourovskaia, A.; Meyer, M.G.; Neumann, T.; Nelson, A.C.; Seibel, E.J. Dual-modal three-dimensional imaging of single cells with isometric high resolution using an optical projection tomography microscope. J. Biomed. Opt. 2009, 14, 064035. [Google Scholar] [CrossRef]
- Staier, F.; Eipel, H.; Matula, P.; Evsikov, A.V.; Kozubek, M.; Cremer, C.; Hausmann, M. Micro axial tomography: A miniaturized, versatile stage device to overcome resolution anisotropy in fluorescence light microscopy. Rev. Sci. Instrum. 2011, 82, 093701. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Bruns, S.; Bruns, T.; Weber, P.; Wagner, M.; Cremer, C.; Schneckenburger, H. Axial tomography in live cell laser microscopy. J. Biomed. Opt. 2017, 22, 91505. [Google Scholar] [CrossRef]
- Weber, M.; Mickoleit, M.; Huisken, J. Multilayer mounting for long-term light sheet microscopy of zebrafish. J. Vis. Exp. 2014, 84, e51119. [Google Scholar] [CrossRef]
- Haghighat, M.B.A.; Akbari, A.; Aghagolzadeh, A.; Seyedarabi, H. A non-reference image fusion metric based on mutual information of image features. Comput. Electr. Eng. 2011, 37, 744–756. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M.; Hübner, B.; Silahtaroglu, A.; Hendzel, M.; Lanctôt, C.; Strickfaden, H.; Cremer, C. The Interchromatin compartment participates in the structural and functional organization of the cell nucleus. BioEssays 2020, 42, 1900132. [Google Scholar] [CrossRef]
- Miron, E.; Oldenkamp, R.; Brown, J.M.; Pinto, D.M.S.; Xu, C.S.; Faria, A.R.; Shaban, H.A.; Rhodes, J.D.P.; Innocent, C.; de Ornellas, S.; et al. Chromatin arranges in chains of mesoscale domains with nanoscale functional topography independent of cohesin. Sci. Adv. 2020, 6, eaba8811. [Google Scholar] [CrossRef]
- Maeshima, K.; Iida, S.; Shimazoe, M.A.; Tamura, S.; Ide, S. Is euchromatin really open in the cell? Trends Cell Biol. 2024, 34, 7–17. [Google Scholar] [CrossRef]
- Gelléri, M.; Chen, S.-Y.; Szczurek, A.; Hübner, B.; Neumann, J.; Kröger, O.; Sadlo, F.; Imhoff, J.; Hendzel, M.J.; Cremer, M.; et al. True-to-scale DNA-density maps correlate with major accessibility differences between active and inactive chromatin. Cell Rep. 2023, 42, 112567. [Google Scholar] [CrossRef]
- von Hase, J.; Birk, U.; Humbel, B.M.; Liu, X.; Failla, A.V.; Cremer, C. Ring Array Illumination Microscopy: Combination of Super-Resolution with Large Field of View Imaging and long Working Distances. bioRxiv 2023. [CrossRef]
- Tsai, P.S.; Mateo, C.; Field, J.J.; Schaffer, C.B.; Anderson, M.E.; Kleinfeld, D. Ultra-large field-of-view two-photon microscopy. Opt. Express 2015, 23, 13833–13847. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. The self-organizing genome: Principles of genome architecture and function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D Nucleome Network. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Ødegård-Fougner, Ø.; Morero, N.R.; Barton, C.; Schueder, F.; Tang, W.; Alexander, S.; Peters, J.-M.; Jungmann, R.; Birney, E.; et al. Nanoscale 3D DNA tracing in single human cells visualizes loop extrusion directly in situ. bioRxiv 2023. [CrossRef]
- Hung, T.-C.; Kingsley, D.M.; Boettiger, A.N. Boundary stacking interactions enable cross-TAD enhancer–promoter communication during limb development. Nat. Genet. 2024, 56, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, S.; Weilandt, E.; Eils, R.; Münkel, C.; Cremer, C.; Cremer, T. Three-dimensional distribution of centromeric or paracentromeric heterochromatin of chromosomes 1, 7, 15, and 17 in human lymphocyte nuclei studied with light microscopic axial tomography. Bioimaging 1995, 3, 121–133. [Google Scholar] [CrossRef]
- Heintzmann, R.; Kreth, G.; Cremer, C. Reconstruction of axial tomographic high resolution data from confocal fluorescence microscopy. A method for improving 3D FISH images. Anal. Cell. Pathol. 2000, 20, 7–15. [Google Scholar] [CrossRef]
- Schneckenburger, H.; Richter, V.; Gelléri, M.; Ritz, S.; Vaz Pandolfo, R.; Schock, F.; von Hase, J.; Birk, U.; Cremer, C. High resolution deep view microscopy of cells and tissues. Quantum Electron. 2020, 50, 2–8. [Google Scholar] [CrossRef]
- Frenkel, N.; Poghosyan, S.; van Wijnbergen, J.W.; van den Bent, L.; Wijler, L.; Verheem, A.; Rinkes, I.B.; Kranenburg, O.; Hagendoorn, J. Tissue clearing and immunostaining to visualize the spatial organization of vasculature and tumor cells in mouse liver. Frontiers 2023, 13, 1062926. [Google Scholar] [CrossRef]
- Lang, F.; Contreras-Gerenas, M.F.; Gelléri, M.; Neumann, J.; Kröger, O.; Sadlo, F.; Berniak, K.; Marx, A.; Cremer, C.; Wagenknecht, H.-A.; et al. Tackling Tumour Cell Heterogeneity at the Super-Resolution Level in Human Colo-rectal Cancer Tissue. Cancers 2021, 13, 3692. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Kaizu, K.; Tamura, S.; Nozaki, T.; Kokubo, T.; Takahashi, K. The physical size of transcription factors is key to transcriptional regulation in chromatin domains. J. Phys. Condens. Matter 2015, 27, 064116. [Google Scholar] [CrossRef] [PubMed]
- Poo, M.; Pignatelli, M.; Ryan, T.J.; Tonegawa, S.; Bonhoeffer, T.; Martin, K.C.; Rudenko, A.; Tsai, L.-H.; Tsien, R.W.; Fishell, G.; et al. What is memory? The present state of the engram. BMC Biol. 2016, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Diederich, B.; Helle, Ø.; Then, P.; Carravilla, P.; Schink, K.O.; Hornung, F.; Deinhardt-Emmer, S.; Eggeling, C.; Ahluwalia, B.S.; Heintzmann, R. Nanoscopy on the Chea(i)p. bioRxiv 2020. [CrossRef]
- Schneckenburger, H.; Richter, V.; Wagner, M. Live-Cell Optical Microscopy with Limited Light Doses; SPIE Spotlight Series; SPIE: Washington, DC, USA, 2018; Volume SL 42, 38p, ISBN 9781510622593. [Google Scholar]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978, 47, 819–846. [Google Scholar] [CrossRef]
- Gibson, G.A.; Loew, L.M. Application of Foerster resonance energy transfer to interactions between cell or lipid vesicle surfaces. Biochem. Biophys. Res. Commun. 1979, 88, 141–146. [Google Scholar] [CrossRef]
- Clegg, R.M.; Sener, M.; Govindjee. From Foerster resonance energy transfer to coherent resonance energy transfer and back, in Optical Biopsy VII (edited by Robert R. Alfano). Proc. SPIE 2010, 7561-12, 75610C. [Google Scholar]
- Masters, B.R. Paths to Förster’s resonance energy transfer (FRET) theory. Eur. Phys. J. 2014, 39, 87–139. [Google Scholar] [CrossRef]
- Schneckenburger, H. Förster resonance energy transfer—What can we learn and how can we use it? Methods Appl. Fluoresc. 2019, 8, 013001. [Google Scholar] [CrossRef]
- Suhling, K.; French, P.M.; Phillips, D. Time-resolved fluorescence microscopy. Photochem. Photobiol. Sci. 2005, 4, 13–22. [Google Scholar] [CrossRef]
- Weber, P.; Schickinger, S.; Wagner, M.; Angres, B.; Bruns, T.; Schneckenburger, H. Monitoring of apoptosis in 3d cell cultures by FRET and light sheet fluorescence microscopy. Int. J. Mol. Sci. 2015, 16, 5375–5385. [Google Scholar] [CrossRef]
- Ma, Y.; Lee, Y.; Best-Popescu, C.; Gao, L. High-speed compressed-sensing fluorescence lifetime imaging microscopy of live cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2004176118. [Google Scholar] [CrossRef] [PubMed]
- Wagnières, G.; Mizeret, J.; Strudzinski, A.; Van den Bergh, H. Frequence-domain fluorescence lifetime imaging for endoscopic clinical cancer photodetection: Apparatus design and preliminary results. J. Fluoresc. 1997, 7, 75–83. [Google Scholar] [CrossRef]
- Squire, A.; Verveer, P.J.; Bastiaens, P.I. Multiple frequency fluorescence lifetime imaging microscopy. J. Microsc. 2000, 197, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 2008, 94, L14–L16. [Google Scholar] [CrossRef] [PubMed]
- Herman, P.; Maliwal, B.P.; Lin, H.-J.; Lakowicz, J.R. Frequency-domain fluorescence microscopy with the LED as a light source. J. Microsc. 2001, 203, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Holst, G.; Gratton, E. Modulated CMOS camera for fluorescence lifetime microscopy. Microsc. Res. Tech. 2015, 78, 1075–1081. [Google Scholar] [CrossRef]
- Mitchell, C.A.; Poland, S.P.; Seyforth, J.; Nedbal, J.; Gelot, T.; Huq, T.; Holst, G.; Knight, R.D.; Ameer-Beg, S.M. Functional in vivo imaging using fluorescence lifetime light-sheet microscopy. Opt. Lett. 2017, 42, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Fruhwirth, G.O.; Ameer-Beg, S.; Cook, R.; Watson, T.; Ng, T.; Festy, F. Fluorescence lifetime endoscopy using TCSPC for the measurement of FRET in live cells. Opt. Express 2010, 18, 11148–11158. [Google Scholar] [CrossRef]
- Becker, W. Fluorescence lifetime imaging—Techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef]
- Becker, W.; Shcheslavskiy, V.; Rück, A. Simultaneous Phosphorescence and Fluorescence Lifetime Imaging by Multi-Dimensional TCSPC and Multi-Pulse Excitation. Adv. Exp. Med. Biol. 2017, 1035, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Bower, A.J.; Li, J.; Chaney, E.J.; Marjanovic, M.; Spillman, D.R., Jr.; Boppart, S.A. High-speed imaging of transient metabolic dynamics using two-photon fluorescence lifetime imaging microscopy. Optica 2018, 5, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, E. Axialtomographie Mit Visualisierung Mikroskopischer Aufnahmen. Bachelor‘s Thesis, Aalen University, Aalen, Germany, 2022. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneckenburger, H.; Cremer, C. Axial Tomography in Live Cell Microscopy. Biophysica 2024, 4, 142-157. https://doi.org/10.3390/biophysica4020010
Schneckenburger H, Cremer C. Axial Tomography in Live Cell Microscopy. Biophysica. 2024; 4(2):142-157. https://doi.org/10.3390/biophysica4020010
Chicago/Turabian StyleSchneckenburger, Herbert, and Christoph Cremer. 2024. "Axial Tomography in Live Cell Microscopy" Biophysica 4, no. 2: 142-157. https://doi.org/10.3390/biophysica4020010
APA StyleSchneckenburger, H., & Cremer, C. (2024). Axial Tomography in Live Cell Microscopy. Biophysica, 4(2), 142-157. https://doi.org/10.3390/biophysica4020010