
Heparin in Acid and Alkaline Environments—A Study of the Correlations between Hydrodynamic Properties and Desulphation

 , , ,
, , , 
 and
and 
Abstract
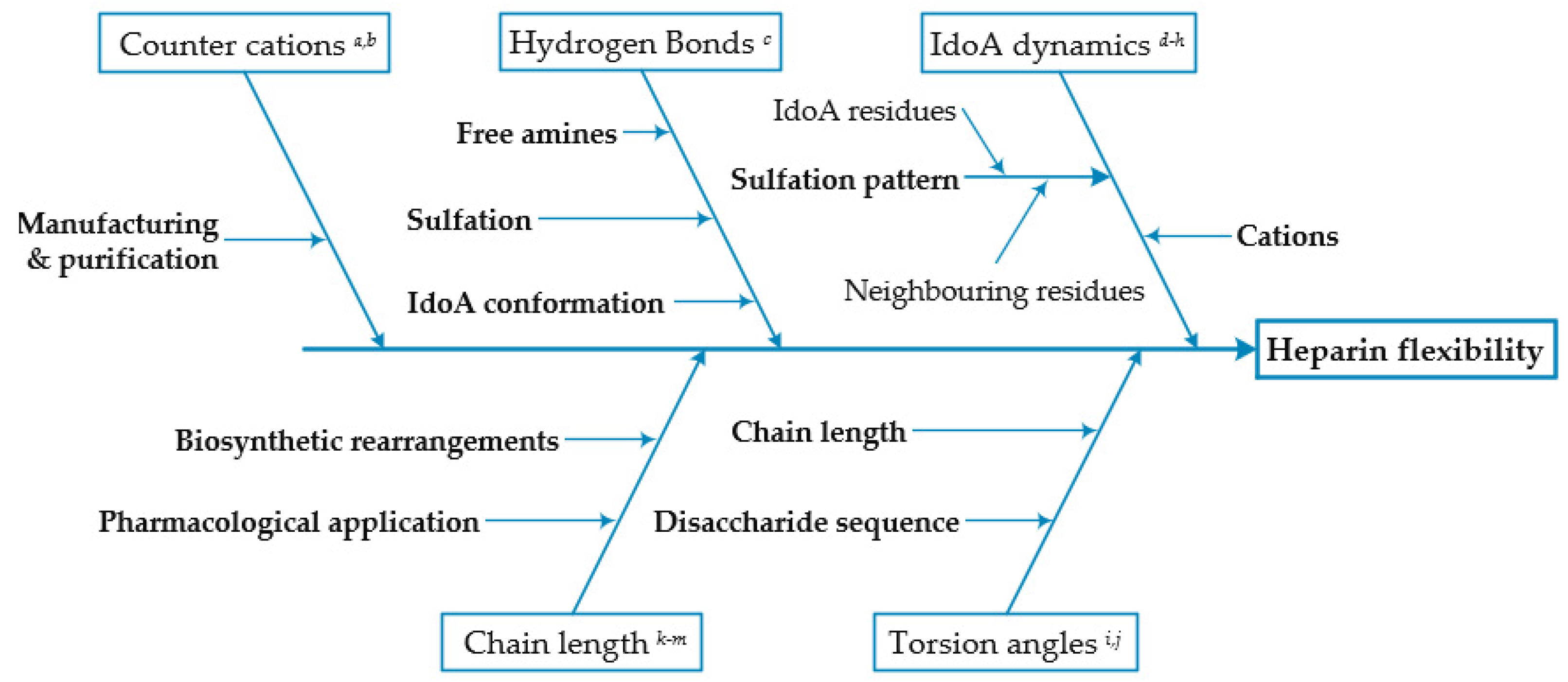
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Solutions
2.3. Heparin Hydrolysis
2.4. Analytical Methods
2.4.1. Size-Exclusion Chromatography
2.4.2. Sedimentation Velocity in the Analytical Ultracentrifuge
2.4.3. Pharmacological Activity Assay
3. Results and Discussion
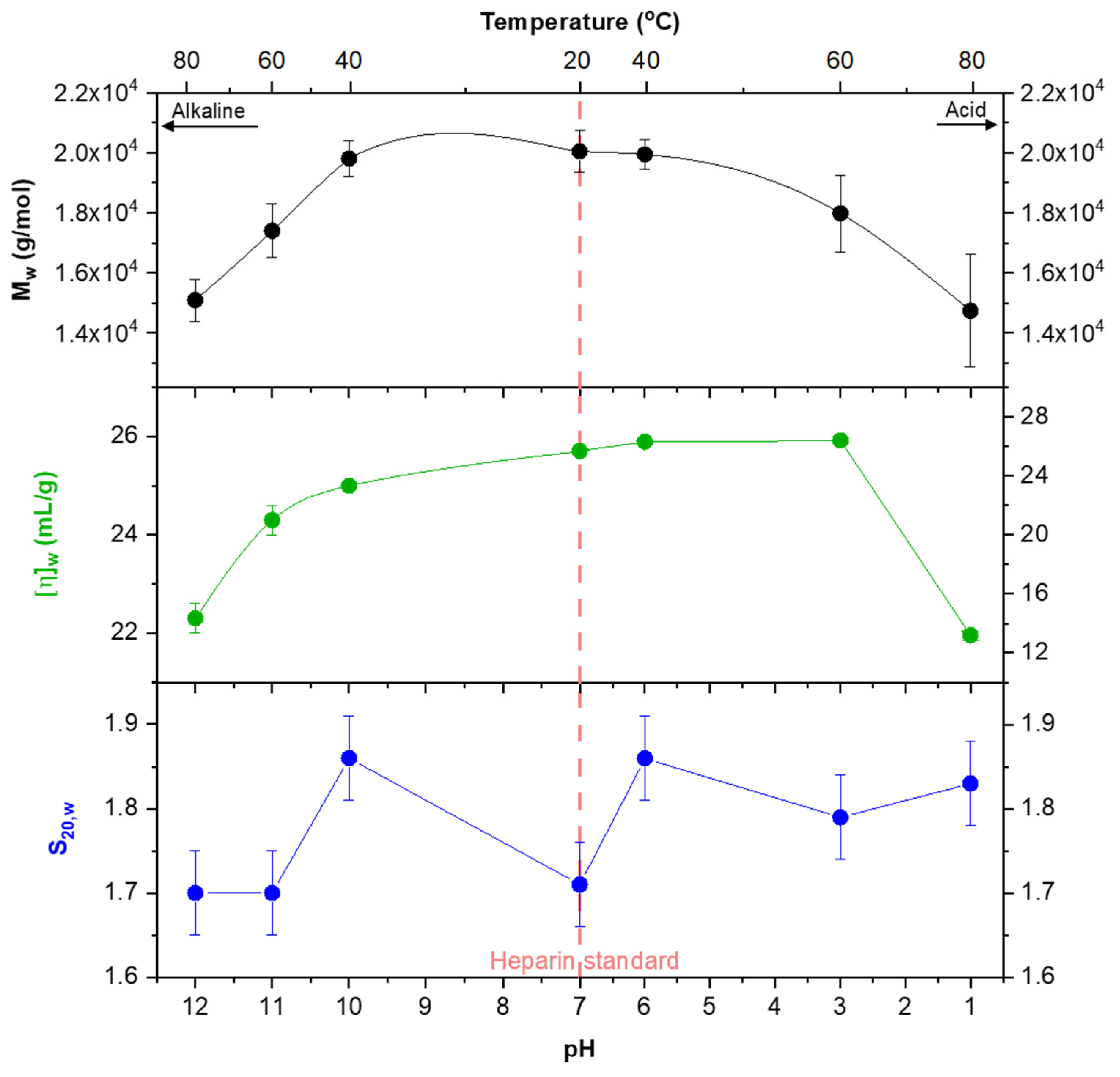
3.1. Analysis with Size-Exclusion Chromatography
3.2. Results of Analytical Ultracentrifugation
3.3. Conformational Analyses
3.3.1. Mark–Houwink–Kuhn–Sakurada (MHKS) Relations
3.3.2. Translational Frictional Ratio
3.3.3. Estimation of Persistence Length
3.4. Activity Change in Light of Hydrodynamic Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mulloy, B.; Khan, S.; Perkins, S.J. Molecular Architecture of Heparin and Heparan Sulfate: Recent Developments in Solution Structural Studies. Pure Appl. Chem. 2012, 84, 65–76. [Google Scholar] [CrossRef]
- Mulloy, B. Heparin—A Century of Progress. In Handbook of Experimental Pharmacology; Lever, R., Mulloy, B., Page, C.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 207, ISBN 978-3-642-23055-4. [Google Scholar]
- Linhardt, R.J.; Claude, S. Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity. J. Med. Chem. 2003, 46, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, U.; Sterner, E.; Hickey, A.M.; Onishi, A.; Zhang, F.; Dordick, J.S.; Linhardt, R.J. Engineering of Routes to Heparin and Related Polysaccharides. Appl. Microbiol. Biotechnol. 2012, 93, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Rudd, T.R.; Yates, E.A. A Highly Efficient Tree Structure for the Biosynthesis of Heparan Sulfate Accounts for the Commonly Observed Disaccharides and Suggests a Mechanism for Domain Synthesis. Mol. Biosyst. 2012, 8, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L. Heparin and Heparan Sulfate: Structure and Function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef] [PubMed]
- Rubinson, K.A.; Chen, Y.; Cress, B.F.; Zhang, F.; Linhardt, R.J. Heparin’s Solution Structure Determined by Small-Angle Neutron Scattering. Biopolymers 2016, 105, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Skidmore, M.; Guimond, S.; Rudd, T.; Fernig, D.; Turnbull, J.; Yates, E. The Activities of Heparan Sulfate and Its Analogue Heparin Are Dictated by Biosynthesis, Sequence, and Conformation. Connect. Tissue Res. 2008, 49, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Palhares, L.C.G.F.; London, J.A.; Kozlowski, A.M.; Esposito, E.; Chavante, S.F.; Ni, M.; Yates, E.A. Chemical Modification of Glycosaminoglycan Polysaccharides. Molecules 2021, 26, 5211. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-Protein Interactions. Angew. Chemie Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Pomin, V.H. NMR Chemical Shifts in Structural Biology of Glycosaminoglycans. Anal. Chem. 2014, 86, 65–94. [Google Scholar] [CrossRef]
- Yates, E.A.; Gallagher, J.T.; Guerrini, M. Introduction to the Molecules Special Edition Entitled ‘Heparan Sulfate and Heparin: Challenges and Controversies’: Some Outstanding Questions in Heparan Sulfate and Heparin Research. Molecules 2019, 24, 1399. [Google Scholar] [CrossRef] [Green Version]
- Onishi, A.; Ange, K.S.; Dordick, J.S.; Linhardt, R.J. Heparin and Anticoagulation. Front. Biosci. 2016, 21, 1372–1392. [Google Scholar] [CrossRef]
- Jaffer, I.H.; Weitz, J.I. Antithrombotic Therapy. In Rutherford’s Vascular Surgery; Cronenwett, J.L., Wayne Johndton, K., Eds.; Saunders Elsevier: Philadelphia, PA, USA, 2014; pp. 549–566. ISBN 9781455753048. [Google Scholar]
- Lundin, L.; Larsson, H.; Kreuger, J.; Kanda, S.; Lindahl, U.; Salmivirta, M.; Claesson-Welsh, L. Selectively Desulfated Heparin Inhibits Fibroblast Growth Factor-Induced Mitogenicity and Angiogenesis. J. Biol. Chem. 2000, 275, 24653–24660. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2015, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, N.; Habuchi, H.; Nagai, N.; Ashikari-Hada, S.; Kimata, K. 6-O-Sulfation of Heparan Sulfate Differentially Regulates Various Fibroblast Growth Factor-Dependent Signalings in Culture. J. Biol. Chem. 2008, 283, 10366–10376. [Google Scholar] [CrossRef] [Green Version]
- Jorpes, E.; Bergström, S. On the Relationship between the Sulphur Content and the Anticoagulant Activity of Heparin Preparations. Biochem. J. 1939, 33, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. N.m.r. and Molecular-Modelling Studies of the Solution Conformation of Heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef]
- Casu, B.; Naggi, A.; Torri, G. Re-Visiting the Structure of Heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef]
- Sun, S.F. Physical Chemistry of Macromolecules: Basic Principles and Issues, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004; ISBN 9786468600. [Google Scholar]
- Cui, S.W. Food Carbohydrates: Chemistry, Physical Properties, and Applications, 1st ed.; Taylor & Francis: Boca Raton, FL, USA, 2005; ISBN 9780849315749. [Google Scholar]
- Finch, P. Carbohydrates Structures, Syntheses and Dynamics, 1st ed.; Springer Science & Business Media: London, UK, 1999; ISBN 9789048140336. [Google Scholar]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Torri, G.; Casu, B.; Gatti, G.; Jacquinet, J.C.; Sinay, P.; Petitou, M.; Choay, J. Evidence for Conformational Equilibrium of the Sulfated L-Iduronate Residue in Heparin and in Synthetic Heparin Mono- and Oligo-Saccharides: NMR and Force-Field Studies. J. Am. Chem. Soc. 1986, 108, 6773–6778. [Google Scholar] [CrossRef]
- Hricovíni, M. Solution Structure of Heparin Pentasaccharide: NMR and DFT Analysis. J. Phys. Chem. B 2015, 119, 12397–12409. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J. Conformation and Dynamics of Heparin and Heparan Sulfate. Glycobiology 2000, 10, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.H.; Thieker, D.F.; Guerrini, M.; Woods, R.J.; Liu, J. Uncovering the Relationship between Sulphation Patterns and Conformation of Iduronic Acid in Heparan Sulphate. Sci. Rep. 2016, 6, 29602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomin, V.H. Solution NMR Conformation of Glycosaminoglycans. Prog. Biophys. Mol. Biol. 2014, 114, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Mulloy, B.; Barrowcliffe, T.W. Heparin and Low-Molecular-Weight Heparin. Thromb. Haemost. 2008, 99, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suflita, M.; Fu, L.; He, W.; Koffas, M.; Linhardt, R.J. Heparin and Related Polysaccharides: Synthesis Using Recombinant Enzymes and Metabolic Engineering. Appl. Microbiol. Biotechnol. 2015, 99, 7465–7479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhardt, R. Chemical and Enzymatic Methods for the Depolymerization and Modification of Heparin. In Carbohydrates: Synthetic Methods and Applications in Medicinal Chemistry; Suami, T., Ogura, H., Hasegava, A., Eds.; VCH Publishers: Cambridge, UK, 1992; pp. 385–401. ISBN 1-56081-701-1. [Google Scholar]
- Khan, S.; Fung, K.W.; Rodriguez, E.; Patel, R.; Gor, J.; Mulloy, B.; Perkins, S.J. The Solution Structure of Heparan Sulfate Differs from That of Heparin: Implications for Function. J. Biol. Chem. 2013, 288, 27737–27751. [Google Scholar] [CrossRef] [Green Version]
- Nunes, Q.M.; Su, D.; Brownridge, P.J.; Simpson, D.M.; Sun, C.; Li, Y.; Bui, T.P.; Zhang, X.; Huang, W.; Rigden, D.J.; et al. The Heparin-Binding Proteome in Normal Pancreas and Murine Experimental Acute Pancreatitis. PLoS ONE 2019, 14, e0217633. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, A.M.; Yates, E.A.; Roubroeks, J.P.; Tømmeraas, K.; Smith, A.M.; Morris, G.A. Hydrolytic Degradation of Heparin in Acidic Environments: Nuclear Magnetic Resonance Reveals Details of Selective Desulfation. ACS Appl. Mater. Interfaces 2021, 13, 5551–5563. [Google Scholar] [CrossRef]
- Bedini, E.; Laezza, A.; Parrilli, M.; Iadonisi, A. A Review of Chemical Methods for the Selective Sulfation and Desulfation of Polysaccharides. Carbohydr. Polym. 2017, 174, 1224–1239. [Google Scholar] [CrossRef]
- Harding, S.E.; Tombs, M.P.; Adams, G.G.; Paulsen, B.S.; Inngjerdingen, K.T.; Barsett, H. An Introduction to Polysaccharide Biotechnology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2017; Volume 80, ISBN 9781315372730. [Google Scholar]
- Van Der Meer, J.Y.; Kellenbach, E.; Van Den Bos, L.J. From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules 2017, 22, 1025. [Google Scholar] [CrossRef] [Green Version]
- Racine, E. Differentiation of the Low-Molecular-Weight Heparins. Pharmacotherapy 2001, 21, 62S–70S. [Google Scholar] [CrossRef] [Green Version]
- Bienkowski, M.J.; Conrad, H.E. Structural Characterization of the Oligosaccharides Formed by Depolymerization of Heparin with Nitrous Acid. J. Biol. Chem. 1985, 260, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, A.M.; Yates, E.A.; Roubroeks, J.P.; Tømmeraas, K.; Larsen, F.H.; Smith, A.M.; Morris, G.A. Monitoring the Stability of Heparin: NMR Evidence for the Rearrangement of Sulfated Iduronate in Phosphate Buffer. Carbohydr. Polym. 2023, 308, 120649. [Google Scholar] [CrossRef] [PubMed]
- Wojtasz-Mucha, J. Pre-Extraction of Wood Components. Mild Hydrothermal Methods for a Future Materials Biorefinery; Chalmers University of Technology: Gothenburg, Sweden, 2020. [Google Scholar]
- Rej, R.N.; Perlin, A.S. Base-Catalyzed Conversion of the a-l-Iduronic Acid 2-Sulfate Unit of Heparin into a Unit of a-l-Galacturonic Acid, and Related Reactions. Carbohydr. Res. 1990, 200, 437–447. [Google Scholar] [CrossRef]
- Danishefsky, I.; Eiber, H.B.; Carr, J.J. Investigations on the Chemistry of Heparin. I. Desulfation and Acetylation. Arch. Biochem. Biophys. 1960, 90, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Jandik, K.A.; Kruep, D.; Cartier, M.; Linhardt, R.J. Accelerated Stability Studies of Heparin. J. Pharm. Sci. 1996, 85, 45–51. [Google Scholar] [CrossRef]
- Mohan, C. Buffers. A Guide for the Preparation and Use of Buffers in Biological Systems, 3rd ed.; EMD Bioscience: San Diego, CA, USA, 2006. [Google Scholar]
- Preparation of Buffer Solutions. Available online: http://delloyd.50megs.com/moreinfo/buffers2.html (accessed on 3 April 2020).
- Mulloy, B.; Heath, A.; Shriver, Z.; Jameison, F.; Al Hakim, A.; Morris, T.S.; Szajek, A.Y. USP Compendial Methods for Analysis of Heparin: Chromatographic Determination of Molecular Weight Distributions for Heparin Sodium. Anal. Bioanal. Chem. 2014, 406, 4815–4823. [Google Scholar] [CrossRef] [Green Version]
- Theisen, A.; Deacon, M.P.; Johann, C.; Harding, S. Refractive Increment Data-Book: For Polymer and Biomolecular Scientists, 1st ed.; Nottingham University Press: Nottingham, UK, 2000. [Google Scholar]
- Harding, S.E. Analysis of Polysaccharides by Ultracentrifugation. Size, Conformation and Interactions in Solution. In Polysaccharides I; Springer: Berlin/Heidelberg, Germany, 2005; Volume 186, pp. 211–254. ISBN 3-540-26112-5. [Google Scholar]
- Schuck, P. Sedimentation Analysis of Noninteracting and Self-Associating Solutes Using Numerical Solutions to the Lamm Equation. Biophys. J. 1998, 75, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.A.; Castile, J.; Smith, A.; Adams, G.G.; Harding, S.E. The Kinetics of Chitosan Depolymerisation at Different Temperatures. Polym. Degrad. Stab. 2009, 94, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.A.; Adams, G.G.; Harding, S.E. On Hydrodynamic Methods for the Analysis of the Sizes and Shapes of Polysaccharides in Dilute Solution: A Short Review. Food Hydrocoll. 2014, 42, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, A.M. Study of Hydrolytic Degradation of Heparin in Acidic and Alkaline Environments; University of Huddersfield: Huddersfield, UK, 2021. [Google Scholar]
- Pavlov, G.; Finet, S.; Tatarenko, K.; Korneeva, E.; Ebel, C. Conformation of Heparin Studied with Macromolecular Hydrodynamic Methods and X-Ray Scattering. Eur. Biophys. J. 2003, 32, 437–449. [Google Scholar] [CrossRef]
- Morris, G.A.; Foster, T.J.; Harding, S.E. A Hydrodynamic Study of the Depolymerisation of a High Methoxy Pectin at Elevated Temperatures. Carbohydr. Polym. 2002, 48, 361–367. [Google Scholar] [CrossRef]
- Tanford, C. Physical Chemistry of Macromolecules; Gaylord, N.G., Gibbs, J.H., Eds.; John Wiley & Sons: New York, NY, USA, 1961. [Google Scholar]
- Harding, S.E. The Intrinsic Viscosity of Biological Macromolecules. Progress in Measurement, Interpretation and Application to Structure in Dilute Solution. Prog. Biophys. Mol. Biol. 1997, 68, 207–262. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; García de la Torre, J. Equivalent Radii and Ratios of Radii from Solution Properties as Indicators of Macromolecular Conformation, Shape, and Flexibility. Biomacromolecules 2007, 8, 2464–2475. [Google Scholar] [CrossRef]
- Morris, G.A.; Ralet, M.-C. The Effect of Neutral Sugar Distribution on the Dilute Solution Conformation of Sugar Beet Pectin. Carbohydr. Polym. 2012, 88, 1488–1491. [Google Scholar] [CrossRef] [Green Version]
- Stivala, S.S.; Yuan, L.; Ehrlich, J.; Liberti, P.A. Physicochemical Studies of Fractionated Bovine Heparin. III. Some Physical Parameters in Relation to Biological Activity. Arch. Biochem. Biophys. 1967, 122, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.; Stivala, S.S. Macromolecular Properties of Heparin in Dilute Solution: 2. Dimensional Parameters as a Function of PH, Ionic Strength and Desulphation. Polymer 1974, 15, 204–210. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Buffering System | Formula | pH Range |
|---|---|---|
| Hydrochloric Acid— Potassium Chloride [45] | HCl + KCl | 1.0–2.2 |
| Citrate Buffer [45] | C6H5O7H2 + C6H5O7Na2·2H2O | 3.0–6.2 |
| Borax Buffer [46] | Na2B4O7·10H2O + NaOH | 9.2–10.8 |
| Phosphate Buffer [46] | NaH2PO4 + NaOH | 10.9–12.0 |
| Sample | Sulphate Removed (%) | Reference |
|---|---|---|
| Heparin Standard | <LOQ *,1 | [53] |
| pH 6/48 h/40 °C | <LOQ * | [34] |
| pH 10/48 h/40 °C | <LOQ * | [40] |
| pH 3/48 h/60 °C | 5.4 (0.8) | [34] |
| pH 11/48 h/60 °C | <LOQ * | [40] |
| pH 12/48 h/80 °C | 14.5 (1.4) | [40] |
| pH 1/48 h/80 °C | 59.8 (1.6) | [34] |
| Mw | [η]w | s20,w | D20,w 107 | Rh | f/f0 | Lp | ML | Lp/ML | a | AntiXa 1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | (g/mol) | (mL/g) | (S) | (cm2s−1) | (nm) | [η] | (nm) | (g/mol.nm) | (mol.nm2/g) | (IU/mL) | |
| Heparin Standard | 20,000 (700) a | 25.7 (0.1) a | 1.71 (0.05) | 4.37 (0.56) | 4.62 (0.28) | 2.8 | 6 | 559 (1) a | 0.0107 | 1.02 | 911 (10) a |
| pH 6/48 h/40 °C | 19,900 (500) a | 26.3 (0.1) a | 1.86 (0.05) | 4.54 (0.37) | 4.54 (0.18) | 2.9 | 6 | 559 (1) a | 0.0107 | 1.02 | 509 (5) d |
| pH 10/48 h/40 °C | 19,800 (600) a | 25.0 (0.1) a | 1.86 (0.05) | 4.59 (0.40) | 4.48 (0.19) | 2.8 | 5 | 559 (1) a | 0.0089 | 0.95 | 723 (8) b |
| pH 3/48 h/60 °C | 17,900 (1300) a | 26.4 (0.2) a | 1.79 (0.05) | 4.77 (0.31) | 4.36 (0.14) | 2.9 | 6 | 548 (2) b | 0.0109 | 1.03 | 266 (6) f |
| pH 11/48 h/60 °C | 17,400 (900) a | 24.3 (0.3) a,b | 1.70 (0.05) | 4.81 (0.46) | 4.26 (0.19) | 2.7 | 6 | 559 (1) a | 0.0107 | 1.04 | 682 (1) c |
| pH 12/48 h/80 °C | 15,100 (700) a,b | 22.3 (0.3) b | 1.70 (0.05) | 5.34 (0.34) | 3.89 (0.12) | 2.7 | 6 | 529 (3) c | 0.0113 | 1.04 | 322 (1) e |
| pH 1/48 h/80 °C | 14,600 (1900) b | 13.2 (0.3) c | 1.83 (0.05) | 6.22 (0.63) | 3.29 (0.16) | 2.3 | 2 | 437 (3) d | 0.0046 | 0.50 | <1 g |
| Pavlov et al. [54] | 4000–37,000 | 8.0–40 | 1.30–3.18 | 3.95–15.40 | 1.39–5.42 | 1.6–3.0 | 3–6 | 570 (50) | 0.0061–0.0105 | 0.90 (0.06) | n/a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozlowski, A.M.; Dinu, V.; MacCalman, T.; Smith, A.M.; Roubroeks, J.P.; Yates, E.A.; Harding, S.E.; Morris, G.A. Heparin in Acid and Alkaline Environments—A Study of the Correlations between Hydrodynamic Properties and Desulphation. Polysaccharides 2023, 4, 88-98. https://doi.org/10.3390/polysaccharides4020007
Kozlowski AM, Dinu V, MacCalman T, Smith AM, Roubroeks JP, Yates EA, Harding SE, Morris GA. Heparin in Acid and Alkaline Environments—A Study of the Correlations between Hydrodynamic Properties and Desulphation. Polysaccharides. 2023; 4(2):88-98. https://doi.org/10.3390/polysaccharides4020007
Chicago/Turabian StyleKozlowski, Aleksandra Maria, Vlad Dinu, Thomas MacCalman, Alan Mark Smith, Johannes Peter Roubroeks, Edwin Alexander Yates, Stephen Ernest Harding, and Gordon Alistair Morris. 2023. "Heparin in Acid and Alkaline Environments—A Study of the Correlations between Hydrodynamic Properties and Desulphation" Polysaccharides 4, no. 2: 88-98. https://doi.org/10.3390/polysaccharides4020007
APA StyleKozlowski, A. M., Dinu, V., MacCalman, T., Smith, A. M., Roubroeks, J. P., Yates, E. A., Harding, S. E., & Morris, G. A. (2023). Heparin in Acid and Alkaline Environments—A Study of the Correlations between Hydrodynamic Properties and Desulphation. Polysaccharides, 4(2), 88-98. https://doi.org/10.3390/polysaccharides4020007