
Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis †


Abstract
:1. Introduction
2. Methods
2.1. General
2.2. Computational Procedure
3. Results and Discussion
4. Conclusions
Funding
Conflicts of Interest
References
- Doroszuk, J.; Musiejuk, M.; Ponikiewski, Ł.; Witt, D. Convenient and Efficient Diastereoselective Preparation of Functionalized Z-Alkenyl Sulfides. Eur. J. Org. Chem. 2018, 2018, 6333–6337. [Google Scholar] [CrossRef]
- Riesco-Domínguez, A.; van de Wiel, J.; Hamlin, T.A.; van Beek, B.; Lindell, S.D.; Blanco-Ania, D.; Bickelhaupt, F.M.; Rutjes, F.P.J.T. Trifluoromethyl Vinyl Sulfide: A Building Block for the Synthesis of CF3S-Containing Isoxazolidines. J. Org. Chem. 2018, 83, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Pramanik, M.; Mandal, A.; Mal, P. S−H···π Driven Anti-Markovnikov Thiol-Yne Click Reaction. Asian J. Org. Chem. 2018, 7, 1849–1855. [Google Scholar] [CrossRef]
- Castarlenas, R.; di Giuseppe, A.; Pérez-Torrente, J.J.; Oro, L.A. The Emergence of Transition-Metal-Mediated Hydrothiolation of Unsaturated Carbon–Carbon Bonds: A Mechanistic Outlook. Angew. Chem. Int. Ed. 2013, 52, 211–222. [Google Scholar] [CrossRef]
- Yang, Y.; Rioux, R.M. Highly regio- and stereoselective hydrothiolation of acetylenes with thiols catalyzed by a well-defined supported Rh complex. Chem. Commun. 2011, 47, 6557–6559. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Marra, A. Metal-Catalyzed and Metal-Free Alkyne Hydrothiolation: Synthetic Aspects and Application Trends. Eur. J. Org. Chem. 2014, 2014, 3955–3969. [Google Scholar] [CrossRef]
- Mancebo-Aracil, J.; Casagualda, C.; Moreno-Villaécija, M.A.; Nador, F.; García-Pardo, J.; Franconetti-García, A.; Busqué, F.; Alibés, R.; Esplandiu, M.J.; Ruiz-Molina, D.; et al. Bioinspired Functional Catechol Derivatives through Simple Thiol Conjugate Addition. Chem. Eur. J. 2019, 25, 12367–12379. [Google Scholar] [CrossRef] [PubMed]
- Daniel, R.M.; Josep, S.V.; Juan, M.A. Catechol-Derivative Compounds And Their Use. WO2019025498, 7 February 2019. [Google Scholar]
- Nador, F.; Volpe, M.A.; Alonso, F.; Feldhoff, A.; Kirschning, A.; Radivoy, G. Copper nanoparticles supported on silica coated maghemite as versatile, magnetically recoverable and reusable catalyst for alkyne coupling and cycloaddition reactions. Appl. Catal. A Gen. 2013, 455, 39–45. [Google Scholar] [CrossRef]
- Moglie, Y.; Buxaderas, E.; Mancini, A.; Alonso, F.; Radivoy, G. Amide Bond Formation Catalyzed by Recyclable Copper Nanoparticles Supported on Zeolite Y under Mild Conditions. ChemCatChem 2019, 11, 1487–1494. [Google Scholar] [CrossRef]
- Buxaderas, E.; Mayer, M.G.; Volpe, M.A.; Radivoy, G. Bimetallic Cu-Pd Nanoparticles Supported on Bio-silica as an Efficient Catalyst for Selective Aerobic Oxidation of Benzylic Alcohols. Synthesis 2017, 49, 1387–1393. [Google Scholar]
- Gutierrez, V.; Mascaró, E.; Alonso, F.; Moglie, Y.; Radivoy, G. Direct synthesis of β-ketophosphonates and vinyl phosphonates from alkenes or alkynes catalyzed by CuNPs/ZnO. RSC Adv. 2015, 5, 65739–65744. [Google Scholar] [CrossRef]
- Menéndez, C.; Nador, F.; Radivoy, G.; Gerbino, D. One-step synthesis of xanthones catalyzed by a highly efficient copper-based magnetically recoverable nanocatalyst. Org. Lett. 2014, 16, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, I.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, E.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Grimme, S.J. Accurate description of van der Waals complexes by density functional theory including empirical corrections. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S.J. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.J. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L.J. Effect of the damping function in dispersion corrected density functional theory. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in 1997, 78, 1396. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.J. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinholt, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005.
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Li, H.; Luo, M.; Tao, G.; Qin, S. Theoretical Calculations on the Mechanism of Enantioselective Copper(I)-Catalyzed Addition of Enynes to Ketones. Catalysts 2018, 8, 359. [Google Scholar] [CrossRef]
- Sirijaraensre, J.; Khongpracha, P.; Limtrakul, J. Mechanistic insights into CO2 cycloaddition to propylene oxide over a single copper atom incorporated graphene-based materials: A theoretical study. Appl. Surf. Sci. 2019, 470, 755–763. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Beletskaya, I.P. Alkyne Insertion into the M-P and M-H Bonds (M=Pd, Ni, Pt, and Rh): A Theoretical Mechanistic Study of the C-P and C-H Bond-Formation Steps. Chem. Asian J. 2011, 6, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Wang, Z.; Zhu, J.; Zhao, Y. Mechanistic Insight into the Copper-Catalyzed Phosphorylation of Terminal Alkynes: A Combined Theoretical and Experimental Study. J. Org. Chem. 2014, 79, 6816–6822. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, L.; Moglie, Y.; Dorn, V.; Radivoy, G. Hydrophosphorylation of aliphatic alkynes catalyzed by CuNPs/ZnO for the synthesis of vinyl phosphonates. A DFT study on the reaction mechanism. RSC Adv. 2017, 7, 18707–18713. [Google Scholar] [CrossRef]




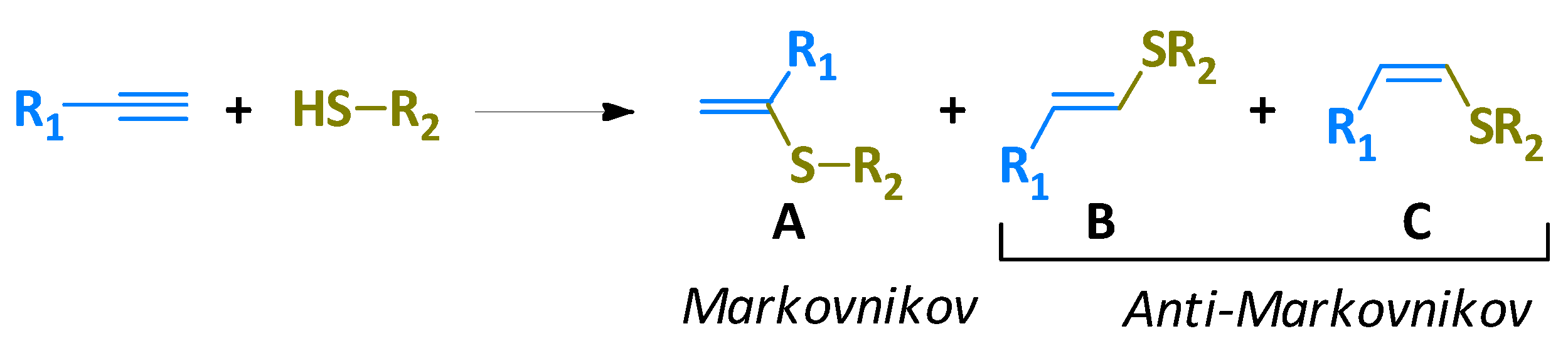{kind=link}
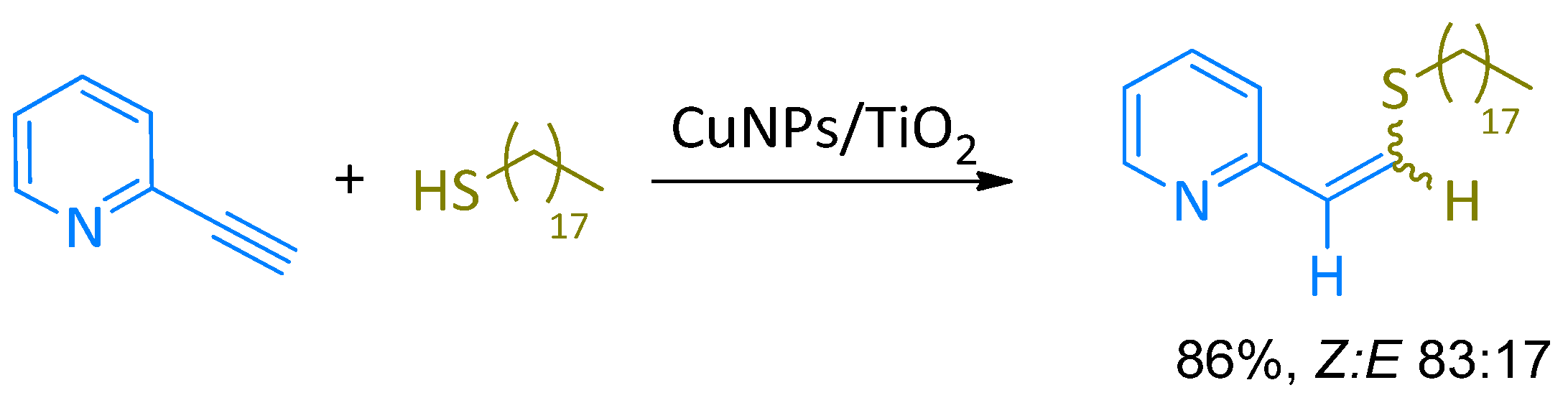{kind=link}
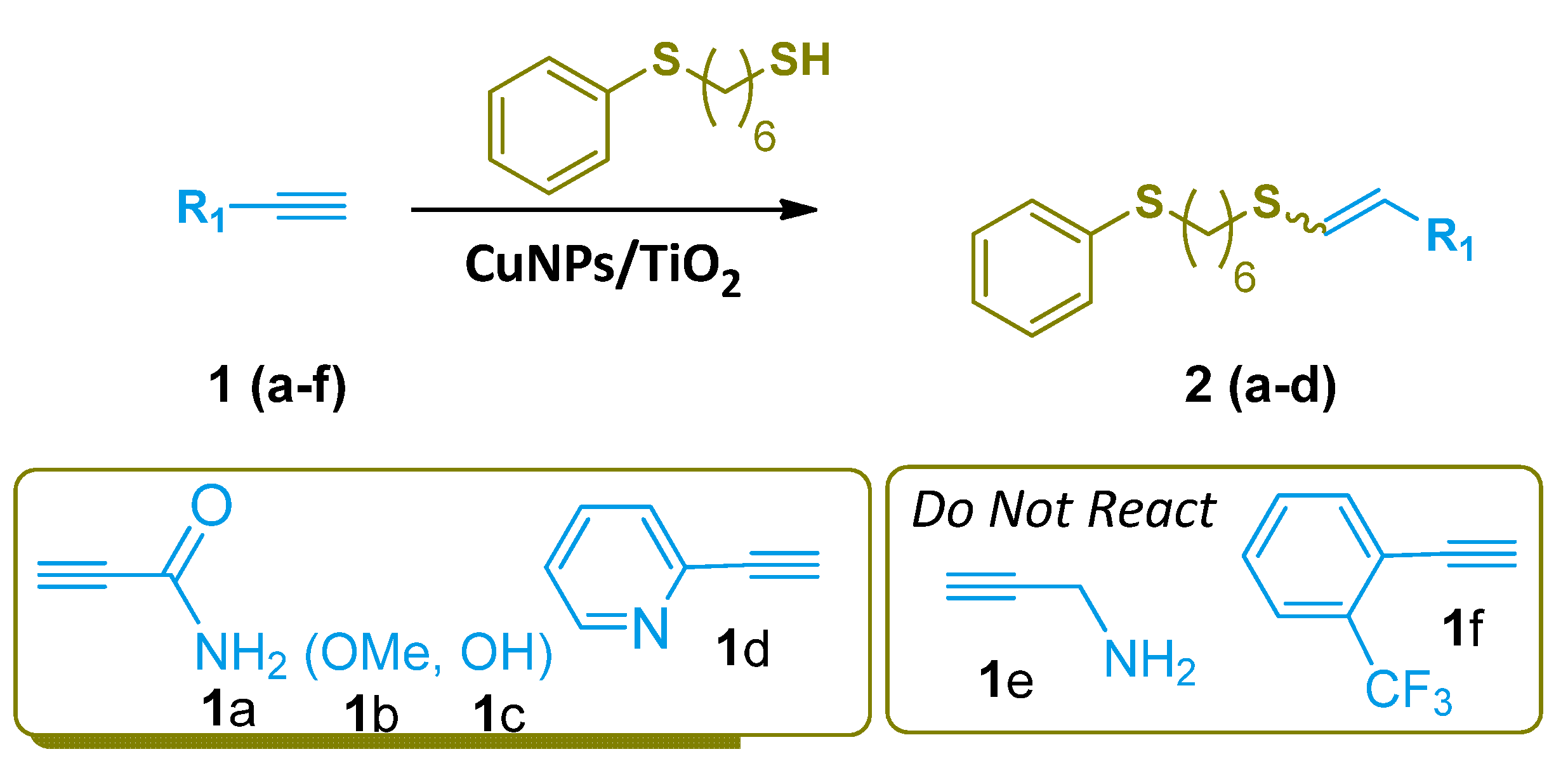{kind=link}
 1a | M06/TZVP(-f) Gas Phase | M06-2X/TZVP(-f) Gas Phase | ||||
| Atom | MULLIKEN | CHELPG | MULLIKEN | CHELPG | ||
| 1 C | −0.098 | −0.303 | −0.135 | −0.324 | ||
| 2 C | −0.299 | −0.165 | −0.075 | −0.153 | ||
| 3 H | 0.223 | 0.286 | 0.192 | 0.303 | ||
| 4 C | 0.726 | 0.863 | 0.464 | 0.842 | ||
| 5 O | −0.524 | −0.551 | −0.465 | −0.544 | ||
| 6 N | −0.470 | −0.980 | −0.436 | −0.997 | ||
| 7 H | 0.219 | 0.423 | 0.229 | 0.432 | ||
| 8 H | 0.223 | 0.428 | 0.225 | 0.442 | ||
| M06-L/TZVP(-f) Gas Phase | M06-L/TZVP(-f) CPCM=DCM | B3LYP/6-311+G** | ||||
| Atom | MULLIKEN | CHELPG | MULLIKEN | CHELPG | NBO Gas Phase | NBO CPCM=DCM |
| 1 C | 0.043 | −0.305 | 0.033 | −0.283 | −0.158 | −0.145 |
| 2 C | −0.329 | −0.157 | −0.339 | −0.219 | −0.105 | −0.124 |
| 3 H | 0.199 | 0.28 | 0.243 | 0.303 | 0.231 | 0.248 |
| 4 C | 0.793 | 0.829 | 0.795 | 0.879 | 0.592 | 0.596 |
| 5 O | −0.630 | −0.533 | −0.739 | −0.647 | −0.574 | −0.643 |
| 6 N | −0.466 | −0.941 | −0.456 | −0.917 | −0.783 | −0.758 |
| 7 H | 0.193 | 0.409 | 0.235 | 0.444 | 0.397 | 0.414 |
| 8 H | 0.197 | 0.418 | 0.228 | 0.44 | 0.401 | 0.414 |
 1b | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.173 | −0.157 | −0.129 | −0.114 | |
| 2 C | −0.329 | −0.387 | −0.106 | −0.131 | |
| 3 H | 0.253 | 0.28 | 0.231 | 0.248 | |
| 4 C | 0.92 | 0.997 | 0.728 | 0.741 | |
| 5 O | −0.523 | −0.602 | −0.552 | −0.583 | |
| 6 O | −0.321 | −0.339 | −0.519 | −0.519 | |
| 7 C | −0.140 | −0.149 | −0.222 | −0.224 | |
| 8 H | 0.127 | 0.148 | 0.189 | 0.198 | |
| 9 H | 0.093 | 0.105 | 0.19 | 0.192 | |
| 10 H | 0.093 | 0.105 | 0.19 | 0.192 | |
 1c | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.238 | −0.216 | −0.124 | −0.098 | |
| 2 C | −0.239 | −0.275 | −0.160 | −0.175 | |
| 3 H | 0.276 | 0.3 | 0.236 | 0.253 | |
| 4 C | 0.78 | 0.857 | 0.724 | 0.739 | |
| 5 O | −0.442 | −0.548 | −0.510 | −0.571 | |
| 6 O | −0.540 | −0.589 | −0.640 | −0.653 | |
| 7 H | 0.403 | 0.471 | 0.481 | 0.505 | |
 1d | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | 0.356 | 0.41 | 0.061 | 0.059 | |
| 2 C | −0.440 | −0.466 | −0.237 | −0.232 | |
| 3 C | 0.22 | 0.244 | −0.167 | −0.160 | |
| 4 C | −0.592 | −0.628 | −0.199 | −0.197 | |
| 5 C | 0.899 | 0.979 | 0.113 | 0.104 | |
| 6 N | −0.638 | −0.765 | −0.430 | −0.465 | |
| 7 H | 0.047 | 0.052 | 0.191 | 0.198 | |
| 8 H | 0.168 | 0.191 | 0.214 | 0.226 | |
| 9 H | 0.078 | 0.095 | 0.212 | 0.224 | |
| 10 H | 0.205 | 0.23 | 0.22 | 0.229 | |
| 11 C | −0.306 | −0.363 | −0.037 | −0.052 | |
| 12 C | −0.253 | −0.239 | −0.167 | −0.176 | |
| 13 H | 0.256 | 0.259 | 0.226 | 0.241 | |
 1e | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.219 | −0.242 | −0.240 | −0.258 | |
| 2 C | −0.385 | −0.416 | −0.036 | −0.036 | |
| 3 C | 0.615 | 0.682 | −0.268 | −0.273 | |
| 4 H | 0.281 | 0.305 | 0.226 | 0.239 | |
| 5 H | −0.018 | −0.022 | 0.214 | 0.221 | |
| 6 N | −1.003 | −1.121 | −0.814 | −0.836 | |
| 7 H | −0.019 | −0.023 | 0.214 | 0.221 | |
| 8 H | 0.374 | 0.418 | 0.352 | 0.361 | |
| 9 H | 0.375 | 0.419 | 0.352 | 0.361 | |
 1f | M06-L/TZVP(-f) | B3LYP/6-311+G** | |||
| Atom | CHELPG Gas Phase | CHELPG CPCM=DCM | NBO Gas Phase | NBO CPCM=DCM | |
| 1 C | −0.325 | −0.356 | −0.157 | −0.170 | |
| 2 C | −0.104 | −0.116 | −0.042 | −0.053 | |
| 3 H | 0.272 | 0.3 | 0.229 | 0.243 | |
| 4 C | 0.26 | 0.241 | −0.107 | −0.115 | |
| 5 C | −0.090 | −0.079 | −0.108 | −0.118 | |
| 6 C | 0.506 | 0.514 | 1.069 | 1.071 | |
| 7 C | −0.110 | −0.122 | −0.176 | −0.175 | |
| 8 H | 0.133 | 0.147 | 0.228 | 0.235 | |
| 9 C | −0.133 | −0.129 | −0.191 | −0.188 | |
| 10 H | 0.127 | 0.144 | 0.213 | 0.224 | |
| 11 C | −0.090 | −0.101 | −0.184 | −0.181 | |
| 12 H | 0.125 | 0.145 | 0.214 | 0.225 | |
| 13 C | −0.188 | −0.187 | −0.152 | −0.153 | |
| 14 H | 0.141 | 0.157 | 0.22 | 0.227 | |
| 15 F | −0.168 | −0.186 | 0.35 | 0.357 | |
| 16 F | −0.184 | −0.185 | −0.358 | 0.358 | |
| 17 F | −0.171 | −0.188 | −0.35 | 0.358 | |
 |  |  |  | |
|---|---|---|---|---|
| Ia | IIa | IIb | IIc | |
| M06-L/TZVP(-f) gas phase | −80 | −228 | −261 | −151 |
| M06-L/TZVP(-f) CPCM=DCM | −74 | −208 | −236 | |
| B3LYP/6-311+G** gas phase | −73 | −208 | −227 | −139 |
| B3LYP/6-311+G** CPCM=DCM | −73 | −207 | −224 |
 | Method | Formation Energy (kcal/mol) |
| M06-L/TZVP(-f), gas phase | −30.4 | |
| M06-L/TZVP(-f), CPCM=DCM | −26.0 | |
| B3LYP/6-311+G**, gas phase | −10.9 | |
| B3LYP/6-311+G**, CPCM=DCM | −30.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capurso, M.; Radivoy, G.; Nador, F.; Dorn, V. Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chem. Proc. 2021, 3, 120. https://doi.org/10.3390/ecsoc-24-08323
Capurso M, Radivoy G, Nador F, Dorn V. Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chemistry Proceedings. 2021; 3(1):120. https://doi.org/10.3390/ecsoc-24-08323
Chicago/Turabian StyleCapurso, Matías, Gabriel Radivoy, Fabiana Nador, and Viviana Dorn. 2021. "Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis" Chemistry Proceedings 3, no. 1: 120. https://doi.org/10.3390/ecsoc-24-08323
APA StyleCapurso, M., Radivoy, G., Nador, F., & Dorn, V. (2021). Synthesis of Alkenyl Sulfides Catalyzed by CuNPs/TiO2. A Theoretical-Computational Analysis. Chemistry Proceedings, 3(1), 120. https://doi.org/10.3390/ecsoc-24-08323